I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
appunti sulla Corrosione AD UMIDO e SULLA Protezione dei materiali METALLICI.
Premessa.
Queste brevi dispense sulla “Corrosione e Protezione dei Materiali Metallici” costituiscono una traccia delle lezioni svolte nell’ambito del Master di Y.E. dell’Università di Pisa. Sono dapprima discussi i fondamenti della corrosione ad umido, poi mostrate le morfologie di danneggiamento più frequenti ed infine discusse le tecniche più adottate di protezione. Come testo di studio e approfondimento si raccomanda lo L.L.Sheir et al. In: “CORROSION” Vol.1 – Vol.2, Eds. Butterworth-Heinemann (1995).
La Corrosione e della Protezione dei Metalli è, per diversi aspetti, una disciplina semiempirica. La risoluzione dei casi di corrosione e l’individuazione dei possibili rimedi sono simili alle diagnosi e terapie medicali in cui, come noto, l’esperienza del ‘tecnico’ può spesso fare la differenza. E’ di fatto assai pericoloso fidarsi della prima impressione che il danneggiamento di un componente può procurare, poiché i fattori che possono giocare un ruolo importante sono molto numerosi ed è alto il rischio di sbagliare la diagnosi finale. Prima di giungere ad una qualsiasi conclusione, occorre reperire quante più informazioni possibili sulla ‘storia’ dell’impianto e sulle condizioni dell’ambiente aggressivo (caratteristiche chimico-fisiche, tipo e condizioni di messa in opera dei materiali ecc.) e condurre tutte le analisi strumentali necessarie per caratterizzare natura, morfologia e dinamica del danneggiamento corrosivo. La corrosione è un campo applicativo multidisciplinare, con basi di Chimica Generale, Elettrochimica e di Metallurgia. La conoscenza della Chimica ed Elettrochimica è necessaria in quanto la corrosione ad umido avviene in ambienti chimici acquosi attraverso reazioni di natura squisitamente elettrochimica. Sulla base di queste considerazioni, nelle dispense sono inseriti alcuni richiami di Termodinamica e Cinetica Chimica/Elettrochimica, così come la classificazione e la Metallurgia delle principali classi di materiali di uso ingegneristico. Vengono infine mostrati e discussi alcuni casi reali di corrosione, al fine di proporre una metodologia di approccio e risoluzione dei problemi.
Con il termine corrosione si indica il complesso dei fenomeni chimico-fisici che comportano il degrado dei materiali metallici ad opera dell'ambiente a cui sono esposti. La corrosione è un lento attacco chimico promosso dall'affinità chimica tra il metallo o lega ed alcuni componenti dell'ambiente (O2, acidi, ecc.). Si parla in tal senso di ANTIMETALLURGIA, poiché il metallo torna alle forme termodinamicamente più stabili, dalle quali era stato sottratto mediante somministrazione di lavoro elettrico o chimico (estrazione, raffinazione).
La maggior parte dei materiali metallici è di fatto TD-nte instabile in ambiente naturale. La spontaneità (e quindi irreversibilità) di una qualsiasi reazione, come ad esempio quella di ossidazione di un metallo (Me° = Mez+ + z e-) in un certo ambiente, può essere stabilita e quantificata dal calcolo dalla diminuzione di Energia Libera di Gibbs associata alla reazione globale che porta all’ossidazione:
DG = DH - TDS (Kcal/mol)
dove DH è la variazione di entalpia associata alla reazione (Kcal/mol), DS la variazione di entropia (Kcal/mol), T la temperatura assoluta in gradi Kelvin.
Se una reazione chimica ha associato un DG <0, vuol dire che la reazione è favorita TD-nte e si svolge irreversibilmente nel verso in cui è scritta. Quando il DG per la reazione ha segno positivo, vuol dire che la reazione non avviene nel verso in cui è scritta ma è spontanea nel verso opposto. Il DG di una reazione ad una data temperatura e con certe concentrazioni (attività) delle specie coinvolte si può calcolare da dati termodinamici tabulati.
Gli ambienti naturali sono in genere caratterizzati dalla presenza di ossigeno e nell’acqua sono sempre presenti ioni H+ che possono, in linea di principio, ossidare molti metalli. Nella Tabella 1.1 si riportano i valori di DG relativi alle reazioni di ossidazione di alcuni metalli con quest’ultime specie. Si osserva come K, Zn e Fe abbiano tutti DG negativi in corrispondenza di entrambe le reazioni, mentre il rame metallico può passare alla forma ossidata solo se presente ossigeno. Infine l’oro metallico non viene ossidato spontaneamente né dall’ossigeno né dall’ambiente acido.
Reazione DG = DH - TDS (Kcal/mol) |
|||
|
Evoluzione H2, (pH = 0) |
Assorbimento O2 (pH =7) |
|
K <-> K+ |
-67.4 |
-86.2 |
|
Zn <-> Zn++ |
-17.9 |
-36.7 |
|
H <-> H+ |
0.0 |
-18.8 |
|
Fe <-> Fe++ |
-11.6 |
-30.4 |
Fe, Zn -> metalli attivi |
Cu <-> Cu++ |
7.8 |
-11.02 |
Cu, Ag -> metalli seminobili |
Au <-> Au++ |
34.5 |
15.7 |
Au, Pt, Pd -> metalli nobili |
Tabella 1.1
Da questi dati si può capire come mai non possono esistere miniere di ferro o zinco metallico, presenti normalmente sotto forma di ossidi/minerali. Esistono invece filoni di oro e platino metallici.
I materiali di uso ingegneristico, quando esposti in atmosfere naturali o industriali, tendono quindi ad ossidarsi, cioé a corrodersi. Quando ciò accade si dice che il materiale è in condizioni di attività. Tuttavia, la condizione termodinamica non è la sola a dover essere considerata. Ha infatti enorme interesse sapere anche a quale velocità procederà il danneggiamento corrosivo.
I danni causati dalla corrosione sono enormi. Il danneggiamento corrosivo spesso non si limita alla semplice sostituzione del componente interessato (costi diretti), ma possono comprendere una serie di danni indiretti, quali ad es. perdite di prodotto attraverso condutture o apparecchiature, fermi impianto non programmati, incidenti agli operatori ecc. Un caso frequente è la perdita di prodotto conseguente alla foratura di un'apparecchiatura di scambio termico; il prodotto entra così nel circuito di raffreddamento e viene scaricato all'esterno con scarse probabilità di individuazione. Danni indiretti sono anche la perdita di efficienza di apparecchiature in conseguenza ad accumuli di prodotti di corrosione; questi fanno aumentare le perdite di carico ed obbligano ad accrescere la potenza di pompaggio, o diminuiscono i rendimenti degli scambi termici modificando i bilanci energetici, le temperature di processo, la qualità e la resa in prodotto finito. Danni indiretti sono da considerare quelli provocati dall'inquinamento dei prodotti (industria alimentare e farmaceutica) e dai conseguenti fermi impianto per permettere la sostituzione delle apparecchiature deteriorate.
Il problema della corrosione deve essere affrontato già in sede di progettazione delle apparecchiature, predisponendo un'oculata scelta dei materiali ed un'efficace sistema di protezione; deve essere inoltre previsto un adeguato sistema di conduzione e manutenzione dell'impianto durante la sua vita operativa seguendo il principio generale di ottenere un grado di affidabilità per i singoli componenti tanto maggiore quanto più vitale è il ruolo da essi ricoperto.
Esistono due tipi principali di fenomeni di corrosione:
a) corrosione a secco, cioè ossidazione dei metalli ad alta temperatura con cinetiche dipendenti dalla Termodinamica Chimica;
b) corrosione ad umido, cioè corrosione in ambiente acquoso attraverso processi di natura elettrochimica dipendenti dalla termodinamica e cinetica elettrochimica.
Nella corrosione ad umido l'ambiente corrosivo è costituito da soluzioni acquose, con funzionamento di sistemi galvanici in cui il processo corrosivo è la risultante di un processo anodico di dissoluzione (ossidazione) del materiale in congiunzione a un parallelo processo catodico di riduzione di una specie presente nell’ambiente acquoso. Queste semireazioni avvengono entrambe in stretta prossimità della superficie metallica.
Consideriamo, ad esempio, il processo di corrosione di una sbarretta di ferro immersa in acqua areata. Il processo corrosivo può essere suddiviso in due processi indipendenti, concomitanti e complementari:
1) processo anodico (ossidazione). Passaggio del ferro in ioni idratati, con un numero equivalente di elettroni "lasciati" sulla superficie del metallo. Fe -> Fe++(nH2O) + 2e-
2) processo catodico (riduzione). Assimilazione dell'eccesso di elettroni da parte di depolarizzatori (atomi, molecole o ioni capaci di essere ridotti al catodo), in questo caso specifico ossigeno molecolare disciolto, tramite la semireazione O2 + 2e- + H2O -> 2OH-
Questi due processi sono indipendenti, avvengono su porzioni differenti della superficie metallica, ma sono comunque strettamente complementari, nel senso che il numero di elettroni nell’unità di tempo lasciati sulla superficie metallica dalla reazione anodica deve essere uguale istante per istante al numero di elettroni nell’unità di tempo consumati dalla reazione catodica [vox = vred]. Se così non fosse, si avrebbe un accumulo di carica elettrica nel metallo.
Queste relazioni avvengono entrambe all’interfaccia metallo-soluzione acquosa e possono comportare modificazioni sensibili dell'ambiente acquoso, come un aumento di pH nelle aree catodiche ed una diminuzione del pH nelle aree anodiche. Possono inoltre avvenire reazioni secondarie quando i prodotti delle differenti semireazioni vengono a contatto fra loro o con altre sostanze presenti nell’ambiente, con separazione ad esempio di ossidi, idrossidi, sali basici ecc.. In questi casi, in funzione della natura e caratteristiche chimico-fisiche degli strati superficiali (continuità, compattezza, conducibilità elettronica, ecc. ecc.) si possono avere sensibili riflessi sulla cinetica degli stessi processi corrosivi.
REAZIONE DI CORROSIONE DEL FERRO IN ACQUA NATURALE
Reazione anodica |
Fe à Fe+++ 2e- |
Reazione catodica |
0.5 O2 + 2e- + H2O à 2OH- |
In soluzione |
Fe++ + 2OH-à Fe(OH)2 |
Reazione globale di corrosione ad umido |
Fe + 0.5 O2 + H2O à Fe(OH)2 |
A causa dell'ambiente esterno l'idrossido ferroso può a sua volta essere ossidato a idrossido ferrico (ma anche direttamente Fe2+àFe3+):
2 Fe(OH)2 + 0.5 O2 à Fe2 O3 . H2O + H2O (Fe2O3 ematite)
Viene così a formarsi la ben nota ruggine di colore rosso-bruno. Tuttavia, in funzione delle caratteristiche ambientali, sono possibili diversi stati di ossidazione del ferro. Di solito, uno strato rugginoso di forte spessore è formato da strati sovrapposti di FeO (Wustite), Fe3O4 .H2O (Magnetite idrata verde), Fe3 O4 (Magnetite Anidra Nera), Fe2O3 .H2O (Ematite rosso-bruna). Il deposito di ruggine è per sua natura discontinuo, poroso e scarsamente protettivo. Sebbene la sua presenza rallenti in qualche misura la velocità di corrosione del metallo sottostante, il danneggiamento prosegue comunque a velocità apprezzabili e dipendenti dall’ambiente corrosivo.
Vi sono anche strati superficiali di prodotti di corrosione abbastanza protettivi, come nel caso dello zinco esposto all'azione atmosferica, dove vengono a formarsi miscugli di ossidi/idrossidi di zinco e vari altri sali basici, capaci di proteggere almeno parzialmente il metallo sottostante; oppure strati spessi e compatti di solfato di ferro per acciai in contatto con acido solforico concentrato.
Nel caso in cui l’interazione tra metallo e ambiente comporti la formazione di composti assai protettivi, capaci cioè di ridurre in modo drastico la velocità di corrosione, si parla di stato di passività del metallo (ad esempio acciai inox, titanio, superleghe base nichel, alluminio in ambienti naturali ).
3- Localizzazione dei processi anodici e catodici. Teoria delle coppie locali e teoria delle tensioni miste.
Il fatto che la corrosione in ambiente umido proceda attraverso un meccanismo di tipo elettrochimico implica che sulla superficie metallica procedano entrambe le semireazioni di ossidazione del metallo e di riduzione di una più specie presenti nell’ambiente acquoso. Molto spesso è possibile distinguere anche visivamente dove siano localizzate le diverse aree elettrodiche. Cioè è possibile identificare quali siano le regioni della superficie metallica dove si sono svolgono le reazioni di riduzione (regioni catodiche) e quelle dove si svolgono le reazioni anodiche (ossidazione del metallo). Un esempio classico è quello della goccia d’acqua depositata sulla superficie di un acciaio, vedi Fig.3.1. In questo caso, come in altri che verranno illustrati ad esempio, si parla di corrosione per areazione differenziale.
Nell’esempio della goccia d’acqua, il processo di corrosione avviene inizialmente sull’intera superficie metallica bagnata, ossidazione del metallo promosso dalla presenza di ossigeno disciolto nell’acqua. La reazione anodica è la dissoluzione del metallo(Fe à Fe+++ 2e-), mentre la reazione catodica è la riduzione dell’ossigeno (0.5 O2 + 2e- + H2O à 2OH-). Il procedere della corrosione determina un impoverimento di ossigeno all’interno dell’intera goccia d’acqua, ossigeno che può essere reintegrato solo per solubilizzazione di questa specie dall’atmosfera esterna e per successiva diffusione attraverso la fase liquida. Il reintegro dell’O2, tuttavia, avviene con relativa facilità nelle regioni più esterne della goccia, mentre in quelle più interne e centrali tende a permanere una concentrazione minore. L’instaurarsi di un gradiente di concentrazione di O2 all’interno della goccia si traduce in un comportamento più catodico della superficie metallica bagnata più prossima all’atmosfera esterna e in un comportamento più anodico di quella al centro della goccia. La regione anodica, dove in prevalenza passa in soluzione il metallo (corrosione), si localizza al centro della goccia, vedi Fig.3.1. Nelle regioni di passaggio tra regioni catodiche e regioni anodiche si incontrano ioni OH- e ioni Fe++ e, superato il prodotto di solubilità dell’idrossido, vengono a precipitare i prodotti di corrosione (Fe(OH)2).
Una evidente separazione tra le aree catodiche e quelle anodiche, così come l’evidenza del passaggio di corrente elettrica dalle prime alle seconde, fu dimostrata da EVANS. Questi immerse una striscia di ferro in un cilindro contenente una soluzione acquosa conduttiva ed osservò, dopo un certo tempo, che l’attacco corrosivo si produceva prevalentemente nelle aree metalliche più lontane dal pelo libero dell’acqua, cioè quelle a contatto con la soluzione più povera di ossigeno disciolto. Evans provò quindi a ritagliare la striscia di ferro lungo la linea di demarcazione tra area corrosa ed area non corrosa, poi riunendo le due striscie con l’interposizione di un isolante ed affidando la connessione elettrica ad un conduttore esterno. In questo circuito inserì un amperometro e rilevò così il passaggio di corrente dalla regione catodica a quella anodica, come in una normale cella galvanica (il flusso di elettroni è, per convenzione, opposto alla direzione di passaggio della corrente).
Si supponga adesso di avere tre vaschette contenenti soluzioni acquose a pH=0 di acido cloridrico (HCl), Fig.3.2. Nella prima vaschetta si immerge una sbarretta di zinco commerciale, nella seconda si immergono due sbarrette di zinco e di ferro saldate tra loro e nella terza una sbarretta di zinco purissimo. Una netta separazione tra regioni a prevalente funzionamento catodico e anodico si può osservare nelle prime due vaschette, mentre nella terza questa separazione non è affatto evidente. Nei primi due casi si osserva lo sviluppo di bollicine di idrogeno gassoso dalla reazione catodica 2H+ + 2e- = H2, che avviene sulle impurezze di ferro presenti nello zinco commerciale nel primo caso e sulla barretta di ferro nel secondo. Tutte le altre aree sono evidentemente sede della reazione anodica di ossidazione dello zinco secondo la reazione Zn = Zn++ + 2e-.
Fig.3.2 – Eterogeneità elettrochimica sulla superficie metallica (caso a e caso b) e perfetta omogeneità (caso c).
In questi primi due casi la natura elettrochimica del processo corrosivo è accertabile visivamente, così come la localizzazione delle aree catodiche/anodiche. Trova diretta giustificazione la cosìdetta teoria delle coppie locali, con cui appare intuitivo schematizzare il sistema corrosivo con una cella galvanica bielettrodica cortocircuitata, in cui la corrente IMN fluisce dal catodo (**) (con superficie data dalla sommatoria di tutte le aree a funzionamento catodico) verso l’anodo (superficie somma della aree anodiche) attraverso l’elettrolita e, vedi Fig.3.3.
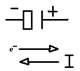 (**) Da un punto di vista elettrochimico il catodo è sempre l'elettrodo sul quale avvengono fenomeni di riduzione. Da un punto di vista elettrico il catodo è sempre l'elettrodo entro cui fluisce corrente proveniente dal conduttore elettrolitico. Il catodo è l’elettrodo positivo (+) quando il sistema galvanico funge da generatore, è l’elettrodo negativo (-) quando il sistema assorbe energia (elettrolisi).
(**) Da un punto di vista elettrochimico il catodo è sempre l'elettrodo sul quale avvengono fenomeni di riduzione. Da un punto di vista elettrico il catodo è sempre l'elettrodo entro cui fluisce corrente proveniente dal conduttore elettrolitico. Il catodo è l’elettrodo positivo (+) quando il sistema galvanico funge da generatore, è l’elettrodo negativo (-) quando il sistema assorbe energia (elettrolisi).
Per contro, nel terzo caso non si riesce a distinguere dove siano le aree catodiche/anodiche, questo perché la superficie della barretta di zinco non presenta alcuna eterogeneità che possa favorire una stabile localizzazione delle reazioni elettrodiche. Si suppone in tal caso che una stessa porzione di superficie possa funzionare da area catodica e da area anodica in un susseguirsi statistico nel tempo. La natura elettrochimica della corrosione si deduce dal fatto che i fattori influenzanti il processo risultante (temperatura, concentrazione delle specie ecc.) agiscono in modo del tutto analogo ai casi precedenti. Secondo questa teoria delle tensioni miste si ribadisce quindi la natura elettrochimica del processo corrosivo anche in caso di assenza di eterogeneità, stabilendo che la velocità di ogni processo parziale anodico o catodico non dipende da quella degli altri processi ma solo dal potenziale e che ad ogni istante la somma delle velocità dei processi anodici uguaglia la somma delle velocità dei processi catodici (assenza di accumulo di cariche). In tali circostanze si può ritenere valida la sovrapponibilità delle curve anodiche e catodiche, come meglio ripreso in seguito.
4 – Superficie elettrificata e doppio strato elettrico. Potenziale assoluto (Galvani) e serie dei potenziali standard riferiti all’elettrodo di idrogeno (SHE).
Supponiamo di immergere una sbarretta di un metallo in una soluzione acquosa. All’inizio un certo numero di atomi metallici passeranno dalla fase solida alla fase acquosa attraverso una reazione di ossidazione Me --- Mez+ + ze- , lasciando sulla superficie del metallo un eccesso di cariche negative che si localizzano sulla superficie metallica all’interfaccia con la soluzione cquosa. Gli ioni metallici positivi entrati in soluzione saranno subito circondati dalle molecole polari dell’acqua (solvatazione). Tuttavia, tenderanno a rimanere in prossimità della superficie metallica a causa dell’attrazione elettrostatica con l’eccesso di carica negativa della superficie. Man mano che aumenta la concentrazione degli ioni metallici passati in soluzione, aumenta anche la probabilità che gli ioni si riprendano gli elettroni in eccesso sulla superficie e si ‘rituffino’ nel reticolo cristallino del metallo attraverso la reazione di riduzione Mez+ + ze- --- Me. Quando la velocità del processo di passaggio in soluzione, cioè la velocità di ossidazione rox, eguaglia la velocità di ritorno nel reticolo cristallino, cioè la velocità di riduzione rred, non si hanno più flussi netti di materia e il metallo si dice in equilibrio termodinamico con la soluzione acquosa. Le condizioni di equilibrio termodinamico non sono condizioni statiche ma dinamiche: si svolge in continuazione una reazione di ossidazione del metallo a dare ioni ed una reazione di riduzione da ioni ad atomi metallici, reazioni che avvengono però alla stessa velocità. Si ha cioé un equilibrio dinamico in condizioni di reversibilità per cui ian = icat = i0 , dove i0 si definisce densità di corrente di scambio. In queste condizioni di equilibrio elettrochimico del metallo (![]() =0) non si ha corrosione, non verificandosi un flusso netto di metallo verso la soluzione acquosa.
=0) non si ha corrosione, non verificandosi un flusso netto di metallo verso la soluzione acquosa.
La distribuzione delle cariche negative sulla superficie del metallo che comunque è presente in condizioni di equilibrio, crea una differenza di potenziale (d.d.p) tra il metallo stesso e la soluzione in cui è immerso, vedi Fig.4.1 Tale d.d.p. è detta TENSIONE GALVANI (MDSj = jM - jS) all’interfaccia metallo / soluzione e rappresenta il POTENZIALE ASSOLUTO dell’elettrodo immerso nel metallo. Il valore di questo potenziale non e’ calcolabile né misurabile. Questo tuttavia non è un problema insormontabile perché, come meglio spiegato nel seguito, si è interessati più a conoscere la differenza di potenziale tra due differenti elettrodi che non il loro potenziale assoluto.
In tali condizioni di equilibrio TD-ico, all’interfaccia metallo/soluzione elettrolitica viene a costituirsi il cosìdetto doppio strato elettrico. La struttura all’interfaccia metallo – soluzione elettrolitica è supposta multistrato:
E’ apparente un’analogia fisica tra la struttura del doppio strato elettrico e una successione di CONDENSATORI in serie (anche se a differenza di quelli reali, la capacitanza del DSE e’ funzione della d.d.p. esistente). (CDSE tipicamente varia tra 10 e 40 mF / cm2 ).
L’importanza del DSE e dell’associato potenziale Galvani MDSj al contatto delle due fasi, e’ notevolissima, essendo presenti campi elettrici al suo interno estremamente elevati, dell’ordine di 107 V / cm.
(*) Ioni di adsorbimento specifico sono ad esempio gli ioni alogenuri (Cl-, F-, Br- ecc). Questi, malgrado la loro carica netta negativa, mostrano una forte tendenza a legarsi con gli atomi metallici della superficie (chemiadsorbimento), anch’essa come vista carica negativamente. La ragione di questa forte tendenza è legata alla presenza di doppietti elettronici sul guscio esterno di valenza degli atomi del VII gruppo della Tavola Periodica, che tendono a riempire gli orbitali 3d insaturi dei metalli di Transizione. Questa tendenza al chemiadsorbimento degli ioni alogenuri è molto importante, poiché spiega la capacità di tali ioni di favorire destabilizzazione e rottura dei film di passività come ossidi/idrossidi dei metalli a comportamento attivo-passivo (vedi prossimi capitoli).
Come già detto, il potenziale assoluto di un metallo immerso in una soluzione elettrolitica non si può misurare né calcolare mediante dati termodinamici. Tuttavia, per i fini di nostro interesse non è importante conoscere il valore dei potenziali assoluti elettrondci, ma piuttosto conoscere la DIFFERENZA tra i valori dei potenziali. Allo stesso modo si ragiona per i potenziali di elettrodo. Si vanno cioé a misurare i potenziali degli elettrodi che interessano (ELETTRO DI DI LAVORO) tutti rispetto ad uno stesso ELETTRODO DI RIFERIMENTO. La differenza di potenziale che misuro per ciascun elettrodo collegato al riferimento lo chiamo Potenziale Elettrico (E) riferito a quel particolare Elettrodo di Riferimento, MDSj - jrif= E.
Una possibile semicella di riferimento è l’elettrodo standard ad idrogeno (SHE), realizzato con una laminetta di platino su cui gorgoglia idrogeno gassoso a P=1,0 atm immersa in una soluzione ad attività unitaria di idrogenioni H+ (praticamente 1 M a pH=0) a 25 °C, vedi Fig.4.2:
Pt(s), H2 (g; P= 1 atm) / H+ (aq; [H+] = 1)
La reazione elettronica risulta 2H+ +2e- ® H2 e ad essa viene assegnato il valore zero per tutte le temperature (STANDARD TERMODINAMICO).
Se adesso colleghiamo un qualsiasi elettrodo (ELETTRODO DI LAVORO) alla semicella standard ad idrogeno (ELETTRODO DI RIFERIMENTO) e misuriamo il valore di f.e.m. della pila corrispondente, si ottiene il Potenziale Elettrodico dell’elettrodo di lavoro riferito all’elettrodo standard di idrogeno E (SHE).
Ad esempio per un certo metallo Me immerso in una soluzione contenente ioni ad attività (assimilata alla concentrazione) [Mez+], il suo POTENZIALE si misurerà realizzando la cella galvanica:
Pt(s), H2 (g; P= 1 atm) / H+ (aq; [H+] = 1) // Mz+ (aq; [Mez+],), Me(s)
f.e.m. = E Me – E (SHE) = EMe – 0,00 = EMe (V)
Quando si considerano tutti elettrodi in condizioni standard (25 °C, concentrazioni delle specie ioniche unitarie (1 M), pressione specie gassose unitarie P=1 atm) si ottiene la SERIE DEI POTENZIALI ELETTRODICI STANDARD (E 0) sulla scala SHE,
Il potenziale elettrodico (E) può calcolarsi anche in condizioni diverse da quelle standard tramite l’equazione di NERST:
1 - Potenziale di riduzione dell’idrogeno in condizioni non standard
2H+ (aq.)+ 2e- ---- H2 (g) E0 =0,00 V SHE
EH+/H2 = E0H+/H2 + (0,0591/2) Log [H+]/PH2 =
E0H+/H2 + (0,0591/2) Log [H+]/1 = – 0,0591 pH (V) SHE
2 – Potenziale riduzione ossigeno in condizioni non standard
O2 (g)+ 4H+ (aq.) + 4e- --- H2O E0=1,23 V SHE
EO2/H2O = E0 O2/H2O + (0,059/4) Log [H+]4/PO2=
E0 O2/H2O + (0,059/4) Log [H+]4/1=
1,23 – 0,0591 pH (V) SHE
3 – Potenziale di riduzione ioni metallici con concentrazione non standard
Mez+ (aq.) + ze- --- Me (s)
EMez+/Me = E0 Mez+/Me + 0,0591/z) Log [Mez+]
4 – Potenziale non standard di una generica reazione redox:
ox + n e- ---- red
Eox/red = E0 ox/red + (0,0591/n)Log (ox/red)
Un processo corrosivo ha luogo quando sono verificati determinati presupposti termodinamici e cinetici. Ad esempio, la reazione tra un metallo M e l'ambiente acquoso contenente ossigeno:
M + 1/2 O2 + H2O = M(OH)2 [4.1]
è TD-mente possibile solo se accompagnata da una diminuzione dell'energia libera DG del sistema, variazione che esprime inoltre il lavoro motore disponibile per il processo stesso. Questa condizione è verificata nel caso del ferro esposto all'atmosfera, vedi Tabella 1.1 e in questo caso si parla di STATO DI ATTIVITA'.
Quando la reazione 4.1 non è TD-mente possibile (DG > 0), il materiale si dice in STATO DI IMMUNITA' (ad es. oro in atmosfera).
Vi sono però casi in cui la reazione è T.D. possibile, ma estremamente lenta dal punto di vista cinetico e si parla allora di stato di PASSIVITA' (è il caso ad esempio del Cr e degli acciai INOX che si ricoprono di un film sottilissimo ma molto protettivo di ossido di cromo).
La reazione generica 4.1 è di natura elettrochimica ed è, come prima visto, il risultato di due reazioni concorrenti, di cui una catodica che porta alla riduzione delle specie accettrici di elettroni (dall'ambiente) e una anodica di dissoluzione delle specie donatrici di elettroni (il metallo). Per comodità si farà riferimento allo schema della cella galvanica bielettrodica cortocircuitata. Considerando le condizioni di circuito aperto della cella galvanica, senza cioè che fluisca corrente elettrica , si ha che:
DG = -zF(f.e.m.) = -zF( Ecateq – Eaneq)
dove: Ecateq / Eaneq sono i potenziali elettrodici di equilibrio, calcolabili mediante l'equazione di Nerst, z = numero di elettroni scambiati, F = costante di Faraday, T = temperatura assoluta in Kelvin.
Si può quindi esprimere la condizione termodinamica affinché la reazione di corrosione possa avvenire:
DG <0 à Ecateq > Eaneq La reazione di corrosione può avvenire (attività), ma bisogna
aggiungere indicazioni sulla velocità del processo (possibile
passività).
DG =0 à Ecateq = Eaneq Reazione in condizioni di equilibrio
DG >0 à Ecateq < Eaneq La reazione di corrosione non può avvenire(immunità).
Le reazioni anodiche che ci interessano sono quelle di dissoluzione dei metalli. Facendo riferimento alla scala dei potenziali standard (25 °C, concentrazione di tutte le specie presenti 1 M, pressione delle specie gassose 1 atm) rispetto all’elettrodo standard di idrogeno (SHE), si può calcolare, ad esempio, il potenziale del ferro in equilibrio con una soluzione acquosa con concentrazione di ioni ferrosi 10-3M (coppia redox Fe++/Fe):
Fe à Fe+++ 2e- reazione anodica
![]()
Se immergiamo una sbarretta di ferro in una soluzione acquosa non contenente ioni ferrosi, per il calcolo del potenziale di equilibrio si può assumere una concentrazione di ioni Fe++ pari a 10-6 Mol/L (ordine di grandezza 0,1 ppm, ai limiti di rilevabilità dello ione con mezzi chimici). In tal modo il potenziale di equilibrio risulta:
E Fe++/Fe = E0 Fe++/Fe + 0,059/2 Log(10-6) = - 0,586 V
Le reazioni catodiche di maggior interesse sono quelle possibili in ambienti naturali (P= 1 atm):
1- Riduzione di O2
O2 + 4H+ + 4e- à 2H2O
![]()
2 - Riduzione di H+ a dare H2h
2H+ + 2e- à H2h
![]()
Nelle soluzioni acquose possono poi esserci altre coppie redox. Ad esempio:
NO2- + 2H+ + e- à NO + H2O
Fe3+ + e- = Fe2+
Cr6+ + 3e- = Cr3+
Pertanto, dalla conoscenza del potenziale elettrodico del metallo nell’ambiente aggressivo d’interesse è possibile dedurre se il metallo è TD-nte o meno aggredibile per corrosione. Basta calcolare il potenziale di equilibrio del metallo (se non sono inizialmente presenti di ioni del metallo si può fare riferimento ad una concentrazione pari a 10-6 M), calcolare il potenziale delle possibili reazioni catodiche presenti in quell’ambiente, e verificare se almeno una di queste è superiore in valore al potenziale di equilibrio del metallo. Se questo accade, allora la reazione di corrosione avviene ed è spontanea. Altrimenti, il metallo è in quell’ambiente in condizioni di immunità termodinamica e non si corrode.
Facendo questo lavoro di confronto dai valori riportati dalla Serie dei Potenziali Standard, si ottengono le seguenti indicazioni di massima:
A) le soluzioni acide disareate possono corrodere tutti i metalli a partire del Pb. (![]() a pH = 0 à 0,0 V SHE);
a pH = 0 à 0,0 V SHE);
B) le soluzioni neutre disareate possono corrodere il Fe e tutti i metalli meno nobili.(![]() a pH =7à-0.414 V SHE);
a pH =7à-0.414 V SHE);
C) Le soluzioni acide areate possono corrodere anche metalli seminobili come l'Ag. ![]() a pH= 0 à +1.23 V SHE);
a pH= 0 à +1.23 V SHE);
D) Le soluzioni contenenti cloro e fluoro possono corrodere tutti i metalli usuali.
Come precedentemente detto, nell’equilibrio di un metallo immerso in una soluzione contenente suoi ioni si svolge in continuazione una reazione di ossidazione del metallo a dare ioni ed una reazione di riduzione da ioni ad atomi metallici, reazioni che avvengono alla stessa velocità ian = icat = i0 . In queste condizioni di equilibrio termodinamico (DG =0) non si ha corrosione del metallo, poiché non c’è un flusso netto di metallo verso la soluzione acquosa. Il potenziale che assume il metallo in queste condizioni è il Potenziale Reversibile di Equilibrio (Eeq.) ,calcolabile, con l'equazione di Nerst:
E Me++/Me = E0 Mez+/Me + (0,0591/z) Log [Mez+]
All'equilibrio metallo-soluzione, il potenziale assunto dal metallo è proprio il potenziale ![]() calcolato con Nerst.
calcolato con Nerst.
In funzione del valore di i0 si possono distinguere i metalli in INERTI (bassa i0 intorno a 1 mA/m2), INTERMEDI (valori di i0 intorno a 10 mA/m2) e normali (alti valori di i0 intorno a 106 mA/m2). Metalli inerti sono Pt, Pd, Fe, Ni, metalli intermedi Cu e Ag, metalli normali Sn, Al, Zn, Pb, Hg.
Se al metallo in condizioni di equilibrio si impone un potenziale di valore più elevato del suo potenziale di equilibrio ![]() , cioè applichiamo una sovratensione anodica ha ad un nuovo valore
, cioè applichiamo una sovratensione anodica ha ad un nuovo valore ![]() ha , il bilanciamento iniziale tra la velocità di ossidazione del metallo e quella di riduzione degli ioni metallici non sarà più rispettato. In particolare, se ci spostiamo con la sovratensione verso valori maggiori di potenziale (più elettropositivi) si favorisce la reazione in senso anodico (OX) e si deprime la velocità di riduzione in senso catodico (il contrario accadrebbe se si fornisse una sovratensione catodica hc che favorirebbe la reazione catodica a scapito della anodica). Si instaura in tal modo un flusso netto di metallo che passa allo stato ossidato in soluzione come ione. Il metallo quindi si consuma, avviene cioè il processo corrosivo del metallo. La sovratensione ha > 0 possiamo imporla dall’esterno collegando il metallo a un alimentatore a corrente continua, oppure la può imporre una reazione redox presente nell’ambiente corrosivo, a patto che abbia un potenziale elettrodico più alto di quello di equilibrio del metallo (Ecat –Ean > 0, quindi DG<0).
ha , il bilanciamento iniziale tra la velocità di ossidazione del metallo e quella di riduzione degli ioni metallici non sarà più rispettato. In particolare, se ci spostiamo con la sovratensione verso valori maggiori di potenziale (più elettropositivi) si favorisce la reazione in senso anodico (OX) e si deprime la velocità di riduzione in senso catodico (il contrario accadrebbe se si fornisse una sovratensione catodica hc che favorirebbe la reazione catodica a scapito della anodica). Si instaura in tal modo un flusso netto di metallo che passa allo stato ossidato in soluzione come ione. Il metallo quindi si consuma, avviene cioè il processo corrosivo del metallo. La sovratensione ha > 0 possiamo imporla dall’esterno collegando il metallo a un alimentatore a corrente continua, oppure la può imporre una reazione redox presente nell’ambiente corrosivo, a patto che abbia un potenziale elettrodico più alto di quello di equilibrio del metallo (Ecat –Ean > 0, quindi DG<0).
Quindi condizione necessaria affinché un metallo si corroda in un ambiente acquoso è che il suo potenziale sia portato ad un valore più elettropositivo (più nobile) del suo potenziale di equilibrio termodinamico in quell’ambiente.
Dato che le correnti elettrodiche corrispondono in pratica a flussi di materia, e la corrente di ossidazione corrisponde alla dissoluzione anodica del metallo, è molto importante sapere come variano tali correnti in funzione delle sovratensioni imposte al metallo. Tali relazioni devono esprimere, in sostanza, l’entità delle dissipazioni energetiche che si accompagnano al procedere delle reazioni elettrodiche. Nel caso delle reazioni elettrochimiche, una prima importante fonte di dissipazione è la sovratensione di attivazione o di trasferimento di carica, associata appunto alla reazione che avviene sull’elettrodo. Questa sovratensione ha lo stesso significato della barriera energetica di energia di attivazione delle reazioni chimiche. L’energia di attivazione in questo caso è associata ai moti di agitazione termica (urti efficaci) e ad una frazione del campo elettrico (a o (1 – a) a seconda che la reazione sia anodica o catodica) nel doppio strato elettrico all’interfaccia metallo-soluzione. L’espressione generale della densità di corrente in funzione della sovratensione è data dall’equazione di BUTLER-VOLMER:
i = i0 ( exp[(1 –a)zFh /RT] – exp[-azFh/RT]
dove a = fattore di simmetria (può variare da 0,3 a 0,7 e d norma si assume pari a 0,5), F = Costante di faraday (96500 C), T = temperatura assoluta). Si può facilmente dimostrare che quando l’entità delle sovratensioni anodiche/catodiche è superiore a 50 mV in valore assoluto, la relazione tra sovratensione e corrente è di tipo logaritmico. In questo caso valgono le equazioni di TAFEL:
per ha > + 50 mV
per hc < - 50 mV
dove ba, bc sono detti coefficienti di Tafel ed i0 è la densità di corrente di scambio. Tutte quest’ultime sono variabili di tipo cinetico.
Si dimostra, parimenti, che nei dintorni del potenziale di equilibrio, cioè per sovratensioni minori in valore assoluto di 30 mV, la relazione tra sovratensione e corrente è di tipo lineare:
dove, Rp= Resistenza di polarizzazione, ia/ic = densità di corrente anodica/catodica.
In un diagramma lineare potenziale-corrente l’andamento complessivo è rappresentato in Fig.6.1. In corrispondenza del potenziale di equilibrio e nei suoi dintorni la relazione h-i è di tipo lineare. Abbastanza lontani dal potenziale di equilibrio (h > 50 mV) la relazione h-i diventa logaritmica (quindi in un diagramma E-i la relazione è rappresentata da una curva). Il ramo con sovratensioni positive (ha) è il ramo di polarizzazione anodica rispetto al potenziale di equilibrio, mentre il ramo con sovratensione negativa (hc) è il ramo di polarizzazione catodica.
Se si utilizza un diagramma semi logaritmico E – log i e riportando in ascissa il valore assoluto della corrente (in modo da avere entrambi i rami della curva di polarizzazione nello stesso quadrante), otterremo nei dintorni del potenziale di equilibrio un andamento non lineare e per ha , |hc | > 50 mV un andamento lineare. Per reazioni bivalenti, come ad esempio per il Fe, si ha che la corrente varia di un fattore 10 ogni 60 mV di sovratensione. Per reazioni monovalenti (riduzione di idrogeno) la variazione invece è di 120 mV/decade.
Nel caso della resistenza per trasferimento di carica, le correnti elettrodiche e quindi la cinetica dei processi elettrodici è descritta dai parametri ba/bc (coefficenti di Tafel) e i0. (densità di corr. di scambio del metallo).
Sovratensione per trasferimento di carica nelle reazioni catodiche di riduzione di idrogeno e ossigeno sui metalli. Si è detto che nei processi corrosivi in ambiente acquoso si ha generalmente, come processo catodico, lo sviluppo di H2 e/o la riduzione di O2 disciolto. Analizziamo più in dettaglio le relative sovratensioni elettroniche.
Per la sovratensione di attivazione H2 si osserva un’anticorrelazione con la sovratensione del metallo con i propri ioni, cioè tanto maggiore è l’ i0,Me quanto minore è l’ i0,H2 su quel metallo.
H3O+ + e- à Hads + H2O
2 Hads à H2
n H2 à n H2h
Il coefficiente di TAFEL![]() costante per i vari materiali è
costante per i vari materiali è ![]() 0,13 / 0,15 (V/decade)
0,13 / 0,15 (V/decade)
La sovratensione di attivazione per l’ O2 sono alte per tutti i metalli (alcune centinaia di mV) e sono funzione del pH e della natura del metallo. Ai bassi valori di corrente catodica le perdite energetiche sono determinate dalla resistenza al trasferimento di carica, ma all’aumentare delle correnti catodiche diventano significative altre perdite energetiche. Infatti, l’O2 disciolto nell’acqua raggiunge la superficie del metallo per diffusione fisica attraverso la soluzione, quindi sotto l’azione di un gradiente di concentrazione e non è facilitata dal trasporto elettroforetico come accde per l’H+. Diventa preponderante un effetto di polarizzazione di concentrazione (![]() ). La resistenza diventa praticamente infinita in corrispondenza di un valore di corrente anodica detta corrente limite
). La resistenza diventa praticamente infinita in corrispondenza di un valore di corrente anodica detta corrente limite
iL
Dove, Co2 = concentrazione O2 nell’ambiente acquoso, D = coefficiente di diffusione O2 nel mezzo acquoso, d = spessore strato limite di Nerst.
7 – Sistema elettrochimico di corrosione. Potenziale misto (di corrosione) e velocità di corrosione.
In un sistema elettrochimico di corrosione si hanno reazioni in cui la corrente (elettroni) passa da una specie chimica ad un’altra del tutto differente (ad esempio dal ferro all’H+) e questo non può essere un processo di equilibrio. Si supponga infatti di avere ferro che si corrode in ambiente acido grazie alla riduzione di ioni H+. In questo caso infatti non si realizza più un bilancio di massa sullo ione metallico, in quanto il processo anodico interessa quasi esclusivamente il metallo (che si corrode), mentre quello catodico interessa quasi esclusivamente l’altra specie coinvolta (H+). Entrambi i processi avvengono però sulla superficie del metallo, ed ognuno avrà un suo potenziale di equilibrio calcolabile con Nerst. In pratica, è come se all'originale equilibrio del metallo immerso in una soluzione contenente suoi ioni, si sia andato a sovrapporre una seconda reazione redox 2H+ + 2e- = H2 che ha un suo potenziale di equilibrio, supposto nell’esempio maggiore (più elettropositivo) di quello del metallo. Il metallo però è un ottimo conduttore elettrico e tenderà ad assumere un solo valore di potenziale. Il potenziale andrà a stabilizzarsi ad un valore di potenziale intermedio tra i due potenziali delle reazioni redox. Si parla quindi di potenziale misto che chiameremo potenziale di corrosione libera del metallo Ecorr. La differenza in valore tra questo potenziale misto e l’originale potenziale di equilibrio della reazione anodica del metallo costituisce la sovratensione imposta alla reazione anodica (ha = Ecorr - Eeq. Fe++/Fe ), e la differenza tra questo potenziale e il potenziale di equilibrio della reazione catodica costituisce la sovratensione imposta alla reazione catodica (hc = Eeq. Me++/Me - Ecorr), tali da far procedere le rispettive reazioni elettrodiche alla stessa velocità (iox = ired). Il potenziale misto Ecorr si stabilizzerà ad un valore per cui tutta la forza motrice disponibile (f.e.m.= Ecat – Ean) verrà dissipata per far avanzare le reazioni elettrodiche e, nel caso ci sia, per vincere la caduta ohmica attraverso la soluzione. Il flusso netto di metallo che passa in soluzione costituisce quindi la corrente anodica e quindi la velocità di corrosione iox= icorr, corrente che si ottiene in corrispondenza del potenziale potenziale di corrosione, Ecorr.Vediamo quindi come operano le due coppie redox indipendentemente tra loro e come si accoppiano a definire le condizioni stazionarie del processo corrosivo del metallo .
Se accoppiamo queste due reazioni redox sulla superficie del metallo e supponiamo che ![]() , si avrà che:
, si avrà che:
Supponendo di operare in soluzione acquosa con conducibilità del mezzo corrosivo molto grande ( RI=0), le condizioni stazionarie di funzionamento si avranno quando tutta la forza motrice disponibile (Ecat – Ean) sarà dissipata nella somma della sovratensione anodica e sovratensione catodica, cioè quando ha + /hc/ = Ecat - Ean (più in generale sarà ha + /hc/ + RI = Ecat - Ean).
Si individuano in tal modo le condizioni stazionarie di funzionamento del sistema corrosivo nel punto di intersezione tra le curve di polarizzazione delle due coppie redox, condizioni che determinano: Ecorr= Potenziale libero di corrosione del metallo (V), icorr = velocità di corrosione ( A/m2)
Il valore del potenziale di corrosione non è calcolabile da dati termodinamici essendo ,come prima detto, associato ad una reazione irreversibile, ma dipenderà essenzialmente da fattori cinetici (valori di i0 e sovratensioni elettroniche ha /hc) legati allo svolgimento dei processi elettronici. Si può dire che le condizioni stazionarie di lavoro del sistema corrosivo si raggiungeranno quando l’intero LAVORO MOTORE disponibile (![]() ) andrà dissipato negli ATTRITI del sistema causati dal passaggio di corrente.
) andrà dissipato negli ATTRITI del sistema causati dal passaggio di corrente.
Cioè: allo stazionario: ![]() = ha + |hc | +RI (con RI caduta ohmica)
= ha + |hc | +RI (con RI caduta ohmica)
NOTA. Nella maggior parte dei casi di corrosione in ambiente acquoso, si tratta di soluzioni ad alta conducibilità ionica e con distanze tra regioni anodiche/catodiche molto ridotte. Pertanto il termine di caduta ohmica RI può essere di norma trascurato e le condizioni di lavoro del sistema corrosivo (Ecorr/Icorr) possono determinarsi dalla intersezione delle curve anodiche e catodiche. Riassumendo:
8 - Diagrammi di EVANS. Teoria delle tensioni miste e sovrapponibilità delle curve di polarizzazione.
Lo studio dei sistemi corrosivi si può efficacemente avvalere della visualizzazione delle curve di polarizzazione anodica e catodica delle reazioni redox coinvolte (Diagrammi di EVANS). A tal riguardo, sono importanti alcune implicazioni derivanti dalla teoria delle tensioni miste. Si è detto che anche in assenza di evidenti eterogeneità superficiali (base della teoria delle coppie locali) la teoria delle tensioni miste ammette la possibilità che su di una stessa porzione di superficie metallica perfettamente omogenea immersa in una soluzione anch’essa omogenea, si possono svolgere contemporaneamente due o più processi elettrodici, alcuni in senso anodico ed altri in senso catodico, in un susseguirsi statistico di posizione e rispetto al tempo. Si possono così riassumere le assunzioni:
In pratica, tale teoria implica la sovrapponibilità delle curve parziali di polarizzazione.
In un qualsiasi sistema corrosivo, anche costituito da più reazioni anodiche e catodiche, le condizioni stazionarie di funzionamento si stabilizzeranno al potenziale Ecorr dove risulterà: ![]() \
\
A titolo d’esempio, vedi Fig. ‘’’’, si può considerare qualitativamente il caso di una sbarretta di ferro immersa in acqua areata a pH = 7. Il potenziale di corrosione libera del metallo Ecorr sarà quel potenziale per cui ![]() (avendo considerato trascurabili le cadute ohmiche attraverso il conduttore elettrolitico).
(avendo considerato trascurabili le cadute ohmiche attraverso il conduttore elettrolitico).
Si intende per condizioni di passività la perdita di reattività del materiale sotto certe condizioni ambientali. La passività è uno stato di drastica riduzione della velocità di corrosione e cioé di scarsa reattività del materiale metallico con l’ambiente aggressivo. Tale stato è in genere associato alla formazione di un film solido interfacciale, molto protettivo e capace di rallentare drasticamente l’attacco corrosivo del metallo sottostante. Gli strati superficiali possono essere costituiti da ossidi o idrossidi, formati quando il metallo o lega è portato a lavorare entro certi intervalli di potenziale superiore a quello di equilibrio (Ecorr > EM/M0 dell’ossido). In questi casi, la modificazione superficiale consiste nella formazione di un film molto sottile (3-5 nm), compatto e protettivo. Oppure si possono formare depositi ben più spessi dei precedenti con prodotti di corrosione o con sali precipitati dalla soluzione acquosa quando si raggiungono le condizioni di superamento dei rispettivi prodotti di solubilità (AgCl, PbSO4, FeSO4, CaCO3 ecc..). Questi strati o film vanno in pratica a costituire una barriera tra il metallo e l’ambiente circostante. Non sempre gli strati superficiali che si formano sulla superficie dei metalli sono protettivi, dipendendo tale proprietà dalla compattezza, porosità, conducibilità ionica ed elettronica. Da un punto di vista ingegneristico il fenomeno è molto importante, e rende conto dell’elevata resistenza alla corrosione di molti metalli e leghe intrinsecamente poco nobili, come Al, Cr, Ti, Ta, e anche acciai inox, superleghe, rame e le sue leghe in acqua di mare, acciaio al carbonio in H2SO4 diluito ecc…
I tipici metalli che esibiscono il fenomeno della passività sono:
Fe, Cr, Ni, Ti, Al e loro leghe.
Nel caso di film sottili di ossidi/idrossidi, la curva di polarizzazione anodica di un materiale con comportamento attivo-passivo esibisce una caratteristica forma a S, vedi Fig. 8.1. Il materiale ha un comportamento attivo (curva definita dai parametri cinetici io e ba) a partire dal suo potenziale di equilibrio fino ad un certo potenziale Epp, detto ‘potenziale di passività primaria’, in corrispondenza del quale inizia la formazione dell’ossido/idrossido.
La regione di passività vera e propria inizia tuttavia in corrispondenza di un potenziale più elettropositivo, detto potenziale di passività Ep, in corrispondenza del quale la densità di corrente anodica scende a valori drasticamente minori (densità di corrente di passività, ip) che permangono quasi invariati per tutto l’intervallo di potenziale di passività. Il potenziale a cui si entra nella regione di passività (Ep) è per molti materiali sensibilmente superiore a (Ecq)M/M0, mentre per metalli nobili come Au e Pt è praticamente coincidente. All’interno dell’intervallo di passività, la densità di corrente anodica rimane grosso modo indipendente dal valore del potenziale del metallo. Le possibili spiegazioni sono due:
Le basse correnti anodiche permangono tali fino ad un potenziale di transpassività ETR, o di sviluppo di ossigeno o di pitting. Al di sopra di questo potenziale intervengono processi anodici concorrenti a quello di dissoluzione del metallo, come ad esempio lo sviluppo di idrogeno, la produzione di ioni del metallo a valenza maggiore o la presenza di ioni cloruro in condizioni sufficientemente ossidanti, cioè per potenziali maggiori ad un certo valore detto di pitting, Epitt, in corrispondenza del quale il film si perfora e dà luogo ad attacco localizzato
La formazione di film di ossidi di passività può essere indotta da condizioni elettrochimiche, come ad esempio nel caso di accoppiamento con un processo catodico (riduzione di O2 e/o scarica di H2 dall’acqua, ad esempio) tale da portare il metallo a lavorare ad un potenziale misto (Ecorr) superiore a quello di formazione dell’ossido (Ecorr > EeqMO), oppure di tipo imposto, quando il processo anodico è stimolato da un potenziale applicato dall’esterno mediante correnti impresse (in questo caso si parla di protezione anodica).
Bisogna sottolineare che non tutti i depositi superficiali o film di ossidi sono fortemente protettivi, in quanto questa proprietà dipende molto dalla continuità, compattezza, conducibilità ionica ed elettronica della barriera formata sulla superficie metallica rispetto all’ambiente corrosivo. Ad esempio, la comune ruggine formata sugli acciai al carbonio è anch’essa un ossido/idrossido, ma non è assai protettiva per la superficie metallica. Di fatto, sono relativamente pochi i casi di depositi superficiali spessi capaci di comportare condizioni di passività. Si possono citare i casi del ferro in acido solforico, ma solo se concentrato (formazione di FeSO4 compatto) oppure del piombo in H2SO4 ma solo diluito, oppure del rame in acqua di mare (Cu2(OH)3Cl). Per contro, sui metalli quali Ti, Fe, Cr, Co, Ni, tutti caratterizzati da una forte affinità per l’ossigeno, cioè ossidabilissimi, si formano film di ossidi estremamente sottili (pochi nm) ma molto continui e compatti, quindi efficaci nel proteggere il metallo sottostante dall’ambiente aggressivo esterno. La natura di questi film non è stata del tutto chiarita. Secondo alcuni autori sono film costituiti prevalentemente da ossidi in diretta corrispondenza con lo strato atomico superficiale, secondo altri sono meglio rappresentabili da uno strato adsorbito di O2.
E’ importante comprendere il perché di un diverso grado di protezione offerto da un film di passività di ossidi rispetto alla comune ruggine formata sul ferro. Entrambi sono a base di ossidi del metallo base ma, mentre i primi sono assai sottili (pochi passi molecolari, al massimo pochi nanometri) e assai protettivi, la comune ruggine non protegge efficacemente il metallo sottostante, che continua a corrodersi. Quel che è diverso è il meccanismo di formazione dei due ossidi. La ruggine, costituita da FeOOH, praticamente ematite idratata, si forma nell’ambiente acquoso o nel film di condensazione presente e poi precipita per superamento del prodotto di solubilità sulla superficie metallica. Il deposito superficiale è per sua natura poco coerente con il substrato metallico e assai poroso; l’ambiente aggressivo raggiunge sempre e comunque il metallo e continua a corroderlo.
Il meccanismo di formazione di un film passivante di ossidi è invece completamente diverso.
In questo caso si può pensare l’ossido superficiale formato direttamente a partire dalla superficie metallica. Il film di ossido è in questo caso coerente e continuo con il metallo, molto protettivo in quanto capace di isolarlo efficacemente dal contatto con l’ambiente aggressivo.
La formazione di questi ossidi avviene più facilmente in soluzioni neutre o basiche. Avviene anche in ambiente acido, dove l’ossido è più instabile, e in questo caso la velocità di dissoluzione dell’ossido determina l’intensità dello stesso fenomeno corrosivo. Ad esempio il cromo (così come gli inox) possono esibire buona passivazione anche in ambienti acidi in quanto gli ossidi di cromo III si dissolvono in modo abbastanza lento
Le variabili ambientali che bisogna considerare attentamente nel valutare l’impiego di un materiale a comportamento attivo-passivo sono:
Il caso di più intersezioni tra la curva anodica del metallo e la curva catodica dell’ambiente (curva 2 in Fig. 8.2) dà lo spunto per spiegare alcuni trattamenti chimici che si impongono ai materiali di molti impianti industriali. Si supponga di fare le seguenti esperienze.
Questi risultati indicano che la passività superficiale è stabile quando si lavora nelle condizioni della curva catodica 2; la curva catodica tipo 1, che corrisponde ad un potenziale redox minore della precedente, può avere più intersezioni con la curva anodica del materiale e quindi sono possibili diverse condizioni di lavoro. Se esponiamo per la prima volta un materiale a comportamento attivo-passivo ad un ambiente dove è attiva la curva catodica 1, le condizioni di lavoro si stabilizzeranno al potenziale di corrosione E corr (1), cioè in condizioni attive del materiale, che potrà quindi corrodersi a velocità apprezzabili. Se invece si è già formato un film di passivazione sulla superficie, questo potrà stabilizzarsi anche in condizioni meno favorevoli: se passo cioè dalle condizioni di lavoro a Ecorr (2) ad un ambiente dove è attiva la curva catodica 1, le condizioni di lavoro si stabilizzeranno questa volta al potenziale di corrosione Ecorr (3), cioè ancora in condizioni di passività.
Questa è la ragione per cui si conducono pre-trattamenti di passivazione nelle apparecchiature nuove prima della definitiva messa in marcia (tubazioni e apparecchiature in rame, in acciai inox ecc). Per gli inox, ad esempio, si immette in circolo una soluzione diluita di acido nitrico o altre soluzioni ossidanti, al fine di rimuovere tutte le impurezze superficiali e formare un film superficiale stabile di passivazione.
Il tasso corrosivo di un materiale metallico è influenzato da tutti quei fattori che possono modificare le specifiche condizioni termodinamiche e cinetiche del processo corrosivo stesso. I principali fattori che concorrono nella definizione del processo elettrochimico possono ricondursi:
La natura e composizione chimica del materiale influenza essenzialmente la nobiltà del processo anodico e l'entità della resistenza offerta nei processi elettrodici. In funzione del particolare accoppiamento metallo-ambiente possono inoltre manifestarsi fenomeni di passività chimica o fisica in relazione all'ambiente ed alle tensioni anodiche raggiunte.
I materiali metallici sono costituiti da un gran numero di cristalli di piccole dimensioni chiamati grani. In seguito ai trattamenti termici o meccanici, questi grani possono assumere orientazioni preferenziali che modificano i valori della polarizzazione e possono facilitare l'innesco di fenomeni corrosivi. Nelle zone di collegamento tra grano e grano vi sono schiere distorte di atomi in posizioni intermedie tra i grani, e quindi tali da risultare in condizioni di maggiore reattività. In tali zone vengono poi a concentrarsi, per effetto di microsegregazione, sensibili quantità di impurezze o anche a prodursi variazioni di composizione chimica a causa di precipitazione di composti intermetallici. Tali zone sono quindi spesso regioni di attacco c. preferenziale (vedi anche la c. intergranulare). Tuttavia, anche all'interno dei grani possono esserci difettosità varie quali difetti puntiformi, lineari (dislocazioni) o di superficie (geminati, stacking faults).
E' noto che imponendo ai materiali metallici deformazioni plastiche a freddo, si possono aumentare caratteristiche meccaniche quali la durezza, carico di snervamento e di rottura. A seguito di questi trattamenti si verificano però anche decrementi della resistenza a c. del materiale. Le ragioni sono molteplici; a livello termodinamico è probabile che vi sia un'influenza sull'attività degli atomi costituenti il materiale metallico (aspetto non ben chiarito), mentre a livello cinetico è ragionevole ipotizzare che un’alta densità di difetti reticolari comporti una maggiore disomogeneità microstrutturale e quindi una maggiore attività elettrochimica. Nel caso di materiali a comportamento attivo-passivo, é anche probabile che gli strati superficiali protettivi siano meno compatti e più difettosi, e quindi meno capaci di svolgere la loro azione schermante. Le sollecitazioni meccaniche indotte da carichi esterni o associate a stati autotensionali, in certe condizioni, possono innescare fenomeni corrosivi assai pericolosi come la Stress-Corrosion-Cracking (S.C.C.), la Hydrogen-Stress-Cracking e la Corrosione-Fatica, di cui si parlerà nel seguito
L'ambiente corrosivo definisce, mediante le sue caratteristiche chimico-fisiche, la natura e nobiltà dei processi elettrodici, ma anche fattori cinetici quali l'entità delle sovratensioni e la stessa possibilità di raggiungere condizioni di passivazione superficiale. Tra le caratteristiche più importanti ricordiamo:
ACIDITA'
L'influenza del pH si esplicita nello stabilire la forza motrice disponibile al processo corrosivo, nonché la possibilità T.D. di precipitazione di ossidi, idrossidi e sali basici, composti che spesso portano alla passivazione del materiale per schermatura. In Fig.9.1 è descritto qualitativamente l'andamento della velocità di attacco di alcuni metalli al variare del pH.
POTERE OSSIDANTE
Il potere ossidante di un ambiente è definito dalla nobiltà del processo catodico che in esso si produce, cioé misurato dal valore della tensione di equilibrio e dall'andamento della relativa caratteristica tensione-corrente. Un aumento di potere ossidante si esplica mediante un incremento della tensione catodica e/o una minore pendenza della curva caratteristica elettrodica. In caso di condizioni generalizzate di attività di un materiale metallico, la disponibilità di un maggior lavoro motore, si traduce in velocità di c. più elevate; tuttavia, nei materiali che esibiscono un comportamento attivo-passivo, tale incremento potrebbe anche comportare il raggiungimento delle condizioni di passivazione e quindi causare un decremento della velocità di c.
TEMPERATURA
L'influenza della temperatura sul fenomeno corrosivo non può essere valutata tout-court, poiché il processo c. è il risultato di processi sia elettrochimici, che chimici (reazioni omogenee) e fisici (diffusione, solubilità). In particolare, la temperatura influisce sulla T.D. della corrosione, sulle sovratensioni (che diminuiscono), sulla conducibilità e sui coefficienti diffusivi (che aumentano), sulla solubilità dei prodotti di c. (che in genere aumenta) e sulla solubilità dei gas disciolti (che diminuisce).
CONDIZIONI DI MOTO RELATIVO
Le condizioni di moto relativo tra soluzione aggressiva e superficie metallica possono influire in vario modo. Nel caso ad es. di un materiale non passivabile in cui la velocità della corrosione è controllata dall'apporto di O2, un aumento della velocità del fluido si traduce in un aumento della velocità di corrosione, v. Fig. 9.2a . Tuttavia nel caso di materiali a comportamento attivo-passivo, nelle stesse condizioni di cui sopra, un maggiore apporto di O2 potrebbe invece determinare l'instaurarsi di una passività generalizzata, vedi Fig.9.2b.
Condizioni di corrosione pericolose possono verificano quando sono presenti condizioni di elevata turbolenza con conseguenti urti, come ad esempio in corrispondenza di brusche variazioni di direzione del fluido, oppure per innesco di abrasione-erosione in presenza di particelle sospese. In questi casi si verifica la cosìdetta corrosione per 'impingment' spesso riscontrata nei tubi degli scambiatori di calore con fluido refrigerante movimentato a velocità eccessiva (corrosione a "dente di sega" delle leghe di rame). La condizione più gravosa si verifica all'imbocco dei tubi in corrispondenza della piastra tubiera, tanto da dover prevedere l'impiego apposite guaine di protezione.
Pericolose sono anche le forti variazioni di pressione, in relazione ai fenomeni di cavitazione che consistono nella rapida formazione e condensazione di bollicine di vapore. Il distacco ed impatto del liquido e del vapore così formati provoca notevole danno erosivo ed anche corrosivo, in quanto viene prontamente distrutto il film protettivo superficiale con formazione di profonde cavità nel metallo. Casi tipici sono le brusche variazioni di pressione all’interno delle pompe e sulle eliche delle navi (incorretto design).
L'attacco corrosivo alle superfici metalliche può essere di tipo diffuso (Corrosione Generalizzata) o può concentrarsi solo su alcune regioni del materiale lasciando le restanti sostanzialmente inalterate (Corrosione Localizzata e Corrosione Selettiva).
Nella Corrosione Generalizzata l’intera superficie del metallo appare danneggiata e la penetrazione del danneggiamento all’interno del materiale può considerarsi grosso modo costante. Si ha di norma quando il materiale lavora in condizioni di attività. In questo caso le aree anodiche e catodiche sono assai piccole e ripartite in modo uniforme sull’intera superficie esposta, senza significative localizzazioni. In queste condizioni, la velocità di perdita di massa per unità di superficie esposta, vm, può essere espressa come:
vm = DW/S t
dove: DW è la perdita di massa (Kg) che si verifica nel tempo t (y), S è la superficie metallica esposta (m2). Se si esprime la perdita di massa in mg, la superficie in dm2 e il tempo in giorni, la perdita di massa è esprimible come unità pratica 1 mdd = 1 mg/(dm2 . day). Può essere utile esprimere la velocità di corrosione come perdita di spessore del componente, vcorr, ad esempio in mm/anno; in tal caso si può usare l’espressione:
vcorr (mm/anno) = 87,6 DW /(g S t)
dove DW = perdita massa (mg), g = densità del metallo (g/cm3), S = superficie esposta (cm2) e t= tempo (ore).
La corrosione generalizzata è di norma meno pericolosa e insidiosa dei fenomeni di corrosione localizzata, in quanto l’entità del danneggiamento è più visibile e quantificabile. Tuttavia, se si verifica in zone nascoste non ispezionabili può anche portare a danni assai gravi, come nel caso di inserti metallici all’interno di elementi lapidei o ceramici. Accade infatti spesso, come nel caso del ferro e degli acciai al C, che i prodotti di corrosione abbiano un volume specifico assai maggiore rispetto al metallo da cui provengono e così nel formarsi determinano forti pressioni interne, che in materiali poco tenaci possono a lesioni e rotture meccaniche del manufatto. La prevenzione per la corrosione generalizzata può farsi in vari modi:
La Corrosione Localizzata può presentarsi con morfologie molto particolari e differenziate a seconda del diverso meccanismo dell'attacco corrosivo; può procedere ad esempio come fenditure o come cricche normali alla superficie del materiale, o può dar luogo a cavità che a seconda della forma assunta sono detti crateri, punte di spillo ecc .
Nella Corrosione Selettiva si ha invece un attacco preferenziale a particolari costituenti strutturali del materiale metallico, o si ha dissoluzione nelle regioni adiacenti il bordo dei grani ecc.
In Figura 10.1 sono riportati alcuni aspetti morfologici tipici dei fenomeni corrosivi. Mentre in condizioni di attacco generalizzato l’entità del danno è proporzionale alla "velocità" di perdita di massa, nel caso di corrosione localizzata tale parametro perde naturalmente ogni significato; in questo caso si preferisce definire la grandezza "intensità" di attacco che tende a dare una misura della massima penetrazione dell'attacco (è in pratica la velocità di penetrazione misurata nel punto di massimo attacco). Naturalmente i valori della velocità e dell'intensità di attacco coincidono nel caso di corrosione uniforme, mentre tendono a divergere per forme di corrosione via via più localizzate; il loro rapporto può quindi essere considerato un indice della localizzazione stessa dell'attacco corrosivo.
I meccanismi di attacco localizzato sono particolarmente pericolosi in quanto possono portare in breve tempo alla perdita di funzionalità di un componente così come dell'intera apparecchiatura, se non ad un vero e proprio cedimento strutturale nel caso di organi sollecitati meccanicamente.
L'attacco corrosivo localizzato è usualmente molto pericoloso sia per l’alta velocità con cui può procedere, sia per la difficoltà stessa di monitoraggio del danno subito dal componente.
Le principali forme di corrosione localizzata sono:
- CORROSIONE PER CONTATTO GALVANICO;
- CORROSIONE PER VAIOLATURA ED IN FESSURA;
- CORROSIONE INTERGRANULARE;
- CORROSIONE PER TURBOLENZA, ABRASIONE, CAVITAZIONE;
- CORROSIONE SOTTO SFORZO;
- CORROSIONE-FATICA.
CORROSIONE PER CONTATTO GALVANICO
Questo tipo di corrosione si verifica quando due o più materiali metallici di diversa nobiltà (Potenziale di Corrosione, Ecorr) sono in contatto elettrico tra loro. L’effetto di accoppiamento può anche verificarsi se uno dei materiali è non metallo ma comunque dotato di una buona conducibilità elettronica (grafite, ossidi o solfuri conduttori, ecc;).
Si supponga di collegare elettricamente due metalli immersi in una soluzione M ed N, di cui M è il metallo meno nobile. Ciascun metallo può essere schematizzato come composto da aree anodiche e catodiche e quindi con un sistema bielettrodico cortocircuitato, vedi Fig. 10.2.
Una volta accoppiati, tra i due sistemi circolerà una corrente I detta corrente di macrocoppia. Tale corrente passa attraverso la soluzione elettrolitica dal materiale meno nobile M a quello più nobile N e va a sovrapporsi alle correnti di microcoppia (coppie locali nei due metalli), facendole variare. Applicando la legge di Kirchhoff al nodo 1 si ha:
Ia = Ic + I
Dove Ia è la corrente anodica, Ic la corrente catodica ed I la corrente esterna di macrocoppia. Risulta quindi che la corrente Ia, cioè la corrente di corrosione di M, aumenta.
Se si applica Kirchhoff al nodo 2 si ottiene:
Ic = Ia + I
Cioè la velocità del processo catodico Ic su N aumenta. In conclusione, l’accoppiamento tra i due metalli di nobiltà diversa comporta un’intensificazione del processo anodico (corrosione) sul metallo meno nobile M e un’intensificazione del processo catodico sul metallo più nobile N.
I fattori principali che influenzano l’entità dell’effetto di accoppiamento galvanico sono:
1 – la differenza di nobiltà pratica tra i due materiali metallici accoppiati;
2 – le proprietà catalitiche del materiale metallico più nobile nei confronti della reazione catodica;
3 – il rapporto tra le superfici dell’accoppiamento che funzionano effettivamente da area anodica e catodica;
4 – la conducibilità elettrica dell’elettrolita.
Differenza di nobiltà pratica. La forza motrice che produce l’attacco galvanico è la differenza di nobiltà pratica dei due metalli a contatto in quello specifico ambiente corrosivo. La nobiltà pratica è in sostanza il potenziale di corrosione libera (Ecorr) che quel metallo assume quando immerso da solo in quel particolare ambiente e, pertanto, varia in ragione dell’ambiente. Ad esempio, un acciaio inossidabile in acqua di acquedotto areata assume una Ecorr elevata di diverse centinaia di millivolt, mentre in acqua deareata e in tutti gli ambienti dove non riesce a passivarsi assume potenziali molto minori e vicini a quello di equilibrio del ferro. In funzione di un cambio di caratteristiche chimico-fisiche di un ambiente si possono anche verificare cambiamenti sostanziali di comportamento corrosionistico dei materiali. Ad esempio, lo zinco è usualmente meno nobile del ferro e normalmente viene corroso nell’accoppiamento e protegge il ferro. Per contro, al di sopra di 60-70 °C si forma un ossido molto protettivo sullo zinco che lo porta a nobiltà maggiori del ferro e nell’accoppiamento è quest’ultimo che si corrode. Lo stagno è normalmente più nobile del ferro, ma quest’ultimo a contatto con alcune sostanze alimentari riesce a passivarsi e diventare più nobile, risultando così protetto per effetto dell’accoppiamento (banda stagnata per i barattoli di conserva).
Una volta specificato l’ambiente è quindi possibile ordinare i metalli secondo il loro potenziale di corrosione libera (nobiltà pratica), come mostrato nella Serie Galvanica dei materiali in acqua di mare.
In genere sono più pericolosi gli accoppiamenti tra metalli molto distanti tra loro nella serie galvanica anche se l'entità dell'attacco può dipendere (anche in maggior misura) dai fattori cinetici che poi intervengono nel processo.
Dissipazioni catodiche. Le proprietà catalitiche del metallo più nobile nei confronti delle reazioni catodiche influenzano l’entità dell’effetto di accoppiamento galvanico sul metallo meno nobile. In generale i metalli in condizioni passive sono meno pericolosi negli accoppiamenti con metalli meno nobili perché, essendo ricoperti da film di ossidi, presentano elevate sovratensioni alle reazioni catodiche e in particolare alla riduzione di ossigeno (più frequente). Ad esempio, tra un accoppiamento inox-alluminio e rame-alluminio in acqua di mare è più pericoloso il secondo perché l’inox, reso passivo dall’accoppiamento, riesce a polarizzarsi più facilmente e, soprattutto, presenta una sovratensione alla riduzione di ossigeno superiore a quella del rame.
Rapporto tra superficie catodica e anodica (Sc/Sa). Un maggior rapporto tra la superficie del metallo più nobile e quello meno nobile esalta l’effetto di corrosione galvanica, risolvendosi in un maggior intensità del danneggiamento di quest’ultimo. Se si immerge una lastra di ferro in acqua con NaCl, il metallo si corroderà ad una certa velocità. Se adesso la stessa lastra si ricopre per metà superficie con una lamina di rame, che praticamente non si corrode e si comporta quindi da catodo, aumenterà l’estensione della superficie catodica disponibile per la riduzione dell’O2 e, dovendo essere necessariamente I red = I ox, aumenterà anche la velocità di corrosione del ferro. Se si ricoprono i 2/3 della superficie della lastra con rame, aumenterà ancora di più l’effetto e, essendosi ridotta la superficie anodica, l’intensità di corrente di corrosione aumenta notevolmente, portando ad una veloce riduzione di spessore del ferro. Da questo semplice esempio si intuisce che nei casi di accoppiamento galvanico ha senso coprire, per esempio mediante pitturazione, la superficie del metallo più nobile e non viceversa. Un esempio applicativo di questo concetto è il caso di un’industria di birra (poco corrosiva) che utilizzava serbatoi in acciaio al C rivestito all’interno con resine fenoliche protettive. Tuttavia, nel rimuovere periodicamente i depositi accumulati sul fondo mediante pale meccaniche, si danneggiava il rivestimento e l’esposizione di ampie superfici metalliche poteva portare a contaminazione del prodotto. Si pensò quindi di placcare il fondo dei serbatoi con un acciaio inox AISI 304, lasciandolo scoperto, continuando a rivestire con pittura le pareti verticali in acciaio al C. Ben presto si presentarono numerose forature passanti sull’acciaio al C nelle regioni adiacenti alla saldatura con l’inox. Gli strati di pitturazione, soprattutto quelli di basso spessore, presentano infatti sempre difettosità e discontinuità, che peraltro aumentano nel tempo. Nel caso citato, l’intera superficie del fondo in inox era disponibile per la riduzione catodica dell’ossigeno e tutta la corrente prodotta andava a concentrarsi in senso anodico su ristrettissime aree di acciaio al C in corrispondenza delle difettosità della pittura. Le elevate densità di corrente anodica producevano pertanto veloci attacchi corrosivi perforanti. La regione più colpita era naturalmente la fascia a ridosso della saldatura con l’inox, in ragione delle minori cadute ohmiche all’interno dell’elettrolita legate alla minor distanza dalle regioni catodiche.
Conducibilità dell’elettrolita. La corrente di macrocoppia che circola tra i due metalli a contatto diminuisce al crescere della resistività dell’elettrolita e, pertanto, decresce l’effetto di accoppiamento galvanico. Ad esempio, la pericolosità degli accoppiamenti bimetallici in acqua dolce a bassa conducibilità è assai più modesto che in acqua di mare, circa 200 volte più conducibile. La Fig.10.3 mostra la distribuzione delle correnti anodiche e catodiche in un accoppiamento zinco-ferro in ambiente acido, mentre la Fig.10.3 mostra come al crescere della resistività dell’elettrolita l’azione galvanica si localizzi sempre più nelle regioni prossime al contatto. Ad esempio pratico:
Come prevenzione dagli effetti di accoppiamenti galvanici si può:
Si diminuisce il pericolo di c. per contatto galvanico scegliendo combinazioni di metalli di nobiltà simile, evitando alti rapporti Scat./San., interponendo isolanti, o anche usando inibitori di corrosione o anodi sacrificabili.
Per "vaiolatura" o "pitting" s'intende una forma di corrosione localizzata con effetto perforante che colpisce i materiali a comportamento attivo-passivo quali il Fe, Al, Cu , Ti, Ni, Cr e loro leghe (in particolare gli acciai inossidabili) quando il potenziale che essi assumono (per effetto della reazione catodica) non è tale da assicurare la formazione di uno strato continuo e protettivo sulla superficie del materiale. Il pitting si verifica in genere in ambienti a carattere ossidante molto debole, contenenti ioni specifici (ad es. cloruri, perclorati, ecc;) oppure in ambienti ossidanti forti (anche qui favorito per presenza di ioni depassivanti). In tal senso, l'acqua di mare risulta un ambiente particolarmente favorevole a questo tipo di corrosione.
Un pit superficiale procede attraverso uno stadio iniziale di innesco e successiva propagazione all’interno del materiale.
L'innesco può verificarsi in corrispondenza dei punti in cui il film passivo è più debole, come in corrispondenza di inclusioni superficiali, difetti del materiale, bande di scorrimento affioranti sulla superficie, variazioni locali dell’aggressività ambientale ecc. Laddove il film viene lacerato, l’ambiente aggressivo raggiunge direttamente il metallo e l’area scoperta diviene anodica rispetto alla regione circostante. Ha luogo quindi un primo danneggiamento localizzato ad opera delle alte densità di corrente anodiche (SC/SA molto grande).
Segue una fase di propagazione, che ha in genere carattere autostimolante ed è dovuto all’istaurarsi di una coppia galvanica tra le aree interne al pit e quelle esterne. In Fig.10.4 è rappresentato il caso di pittino per un metallo esposto ad una soluzione areata contenente NaCl: l'O2 presente nell’acqua viene consumato all’interno del pit e non viene facilmente reintegrato per diffusione dal cuore della soluzione, determinando così una intensificazione della reazione anodica all’interno della cavità. L’aumentata concentrazione degli ioni del metallo nel pit limita peraltro la stessa solubilità dell’ossigeno in soluzione acquosa. Per contro, all’esterno del pit continua a ridursi ossigeno con produzione di ossidrili OH- che favoriscono il permanere della passività nelle regioni circostanti il pit. L’ossigeno stenta a entrare per diffusione nel pit anche per impedimenti geometrici e per locale precipitazione di prodotti di corrosione.
All'interno del pit l’ambiente diviene progressivamente più aggressivo. Questo accade sia per un decremento anche sensibile del pH (idrolisi dello ione metallico M++ per la reazione di idrolisi salina M++ + 2H2O = M(OH)2 + 2H+), sia per possibile trasporto elettroforetico di ioni aggressivi come i Cl- dal bulk della soluzione all’interno del pit. Partendo da una soluzione neutra con poche decine di ppm di ioni cloro nella soluzione aggressiva, si può arrivare con questo meccanismo ad avere pH molto acidi all’interno del pit (pH = 2-3) con tenori di cloruri anche di alcune centinaia di ppm.
Il pitting si verifica tipicamente quando il materiale a comportamento attivo-passivo lavora (a Ecorr) in ambienti troppo poco ossidanti (regione di transizione attivo-passivo) o potenziali elevati, cioè in ambienti molto ossidanti in campo di transpassività. In quest’ultimi, quando Ecorr supera una soglia Epitt si innesca e si propaga la corrosione localizzata. Se invece Ecorr è inferiore di Epitt, il pitting non si verifica.
Gli ioni cloruro, così come in genere gli ioni degli elementi alogenuri (F-, Br- ecc.) tendono a ridurre l’intervallo dei potenziali di passività, cioè ad abbassare Epitt, vedi Fig.10.5 Questo accade perché gli ioni di alugenuri sono ioni di adsorbimento specifico che, come visto hanno una fortissima tendenza a chemiadsorbirsi sugli atomi superficiali del metallo e, ciò facendo, tendono a destabilizzare e facilitare la fessurazione dei film di passività.
Anche un aumento della temperatura o dell’acidità dell’ambiente aggressivo agiscono sfavorevolmente sulla stabilità dei film di passività, semplicemente perché in entrambi i casi aumenta la solubilità degli ossidi/idrossidi superficiali. L’effetto di tali variabili è qualitativamente lo stesso di un aumento della concentrazione di cloruri mostrata in Fig. 10.5
In pratica, la resistenza a pitting di un materiale metallico come ad esempio gli acciai inox è legata all’abilità di formare e di mantenere stabile e compatto il film di passività in quello specifico ambiente acquoso. Negli acciai inox, un maggiore contenuto di cromo e l’aggiunta di elementi quali il molibdeno (Mo) e l’azoto (N) risulta benefico nei riguardi della resistenza a pitting del materiale. Una comparazione qualitativa di massima della resistenza a pitting tra gli acciai inox può essere di fatto ottenuta con l’indice ell’indice PREN (Pitting Resistance Equivalent Number), calcolato sulla base della composizione chimica del materiale:
PREN = %Cr + 3,3%Mo + 16%N per acciai inox austenitici
PREN = %Cr + 3,3%Mo + 30%N per acciai inox duplex
In Tabella 10.1 si riportano i valori di PREN per una serie di acciai inox di interesse. Si può notare come un acciaio AISI 316 abbia un valore di PREN superiore a quello di un AISI 304, e questo grazie all’addizione nell’AISI 316 del 2% di molibdeno, elemento capace di contrastare l’azione depassivante ad opera degli ioni cloruro. La maggior qualità dell’AISI 316 si traduce in un aumento di costo pari a circa il 35% rispetto all’AISI 304.
Gli acciai duplex (2205/2207) hanno valori di PREN superiori all’AISI 316 e, in luogo di un certo aumento di prezzo, possono garantire resistenze a pitting e corrosione interstiziale superiori. In particolare, gli acciai duplex mostrano un significativo incremento delle caratteristiche meccaniche ed una resistenza assai maggiore a fenomeni di corrosione localizzata quale la corrosione sotto sforzo (tensocorrosione, Stress Corrosion Cracking). Maggiori resistenze a pitting sono esibite anche dagli inox superaustenitici (256 SMO, lega 904L) e dalle superleghe base nickel (Hastelloy C22) di costo però sensibilmente superiore.
Normativa |
Denomi |
Cr |
Ni |
Mo |
N |
Altri |
PREN |
1.4301 |
AISI 304 |
18 |
9 |
- |
- |
- |
18 |
1.4436 |
AISI 316 |
18 |
10 |
2 |
- |
- |
25 |
X3CrNiMoN 22 5 |
SAF 2205 |
22 |
5,3 |
3 |
0,16 |
- |
35 |
UNS S 32750 |
SAF 2207 |
25 |
7 |
3,8 |
0,28 |
- |
41 |
X3CrNiMoCu 25 6 |
UR 52N |
25 |
6 |
3,8 |
0,26 |
1,5 Cu |
|
1.4529 ecc. |
904L |
20-22 |
20-22 |
5-6 |
0,1-0,2 |
|
> 40 |
Tabella 10.1 – Confronto dei valori di PREN di diverse classi di acciai.
Per comparare la resistenza a pitting dei diversi materiali metallici si possono condurre prove di laboratorio standardizzate. Si impiegano di norma soluzioni aggressive con alto potenziale redox, basso pH e con presenza di cloruri depassivanti, variando la temperatura di prova fino a determinare le condizioni critiche di innesco del pitting. In queste prove si impiega di norma una soluzione aggressiva al 4% di NaCl + 0,1% Fe2(SO4)3 + 0,01 M HCl e si immergono i campioni per 24 ore nella soluzione variando la temperatura di 5 gradi centigradi per volta. L’obiettivo è quello di determinare la cosìdetta temperatura critica di pittino, quando cioè si iniziano ad osservare attacchi corrosivi con un microscopio ottico a 40 X. I provini sono semplici sbarrette del metallo. Per avere un’idea delle differenti temperature critiche nei materiali metallici di maggior impiego si veda la Tabella 10.2. L’informazione tratta da questa prova ha validità di comparazione tra i materiali e non è un dato assoluto. In altre parole, le temperature riportate sono quelle critiche nella soluzione impiegata e che in un diverso ambiente potrebbero essere anche molto differenti, sebbene il ranking tra i materiali sarebbe comunque lo stesso.
LEGA |
Temperatura critica di pittng |
Temperatura critica di c.interstiziale |
Hastelloy C-22 |
> 150 |
102 |
Hastelloy C-276 |
150 |
80 |
Ferralium 255 |
50 |
35 |
Lega 904L |
45 |
20 |
AISI 317L |
25 |
15 |
AISI 316L |
20 |
< -5 |
Tabella 10.2 – Confronto tra temperature critiche di pitting e di corrosione interstiziale per diverse classi di acciai nella soluzione standard del test ASTM.
CORROSIONE INTERSTIZIALE.
La c. interstiziale è una forma di c. localizzata ad azione cavernizzante che insorge in corrispondenza di interstizi o altri punti schermati per i quali risulta difficoltoso il ricambio della soluzione tra le zone più interne e la massa della soluzione. L'ossigeno o le altre specie ossidanti non riescono a raggiungere le zone interne schermate e sostenere lo stato di passività. Il meccanismo di propagazione della corrosione è praticamente lo stesso della c. per pitting, cioè autostimolante. Una volta consumato l’ossigeno disciolto nella soluzione acquosa all’interno dell’interstizio si innesca una corrosione per areazione differenziale, con localizzazione delle regioni anodiche all’interno dell’interstizio, mentre la reazione catodica sostiene per alcalinizzazione la passività sulle superfici esterne, vedi Fig. 10.3. L’immissione di ioni metallici in soluzione all’interno dell’interstizio comporta idrolisi salina e locale decremento del pH; si favoriscono anche incrementi di ioni depassivanti come i cloruri per trasporto elettroforetico dal corpo della soluzione esterna verso le regioni schermate (meccanismo della cella occlusa). La c. interstiziale si verifica più facilmente rispetto alle condizioni di insorgenza del pitting, poiché a differenza di quest’ultimo esistono già condizioni favorevoli per il funzionamento della cella occlusa.
Per comparare la resistenza dei diversi materiali metallici alla c. interstiziale si conducono di norma test di corrosione analoghi a quello prima visto per la determinazione delle temperature critiche di pittino. I provini in questo caso consistono in laminette del materiale su cui si appoggiano strettamente mediante una molla dei cilindretti di teflon allo scopo d creare un interstizio. Si procede come per il pittino, determinando in questo caso la cosìdetta temperatura critica di corrosione interstiziale. In Tabella 10.2 si riportano le temperature critiche ottenibili con la prova standard ASTM. Si può infine notare come le temperature critiche di innesco della c. interstiziale siano sempre minori di quelle di innesco della c. per pitting. Si apprezzi inoltre la forte differenza di resistenza tra un AISI 316 e un Hastelloy tipo C. E’ opportuno sottolineare che questa tabella è utile per un confronto qualitativo tra diversi materiali e che le temperature riportate hanno una stretta attinenza all’ambiente corrosivo impiegato. In altre parole, in acqua di mare, le temperature critiche di innesco pitting per un AISI 316 sono senza dubbio superiori di 20 °C.
La c. intergranulare si verifica in corrispondenza dei bordi di grano, senza agire in maniera apprezzabile sulla matrice dei grani stessi. E' un attacco insidioso poiché può penetrare profondamente nel metallo senza che i prodotti di corrosione risultino visibili sulla superficie esterna. Il distacco dei legami intergranulari comporta ovviamente la degradazione delle proprietà tecnologiche del materiale. Nel caso di metalli o leghe spesso intervengono fattori strutturali di disomogeneità quali l'accumulo di impurezze, segregazione o precipitazione di fasi particolari (sensibilizzazione), che possono creare condizioni favorevoli per una dissoluzione selettiva. Alte velocità di penetrazione possono raggiungersi quando si possono formare microelementi galvanici in corto circuito in cui il bordo del grano diviene anodico (SA) ed il corpo del grano catodico (SC) con SC>>SA, per cui le densità di corrente anodiche possono raggiungere valori assai elevati. In questo caso le condizioni elettrochimiche sono del tutto analoghe a quelle di innesco del pitting. Pertanto è necessario che, al di là dei necessarie condizioni favorevoli da un punto di vista metallurgico, l'ambiente sia solo blandamente aggressivo, in modo da assicurare condizioni di dissoluzione selettiva.
La c. intergranulare colpisce sia metalli che leghe. Ad esempio l'alluminio commerciale che, se trattato a 600 °C, si sensibilizza per precipitazione sui bdg di impurezze (Fe,Si) e subisce questo tipo di attacco se in contatto con HCl o alcali. Le leghe Al-Cu (durallumini) si possono sensibilizzare per precipotazione di CuAl2 ai bdg con impoverimento di rame nelle zone di precipitazione che diventano anodiche rispetto al corpo del grano. Anche gli acciai inox austenitici possono sensibilizzarsi in seguito a trattamenti a temperature tra 400-850 °C anche per tempi brevi. In queste condizioni precipitano dei carburi di cromo (M23C6) sui bdg provocando un impoverimento di cromo libero nelle regioni adiacenti, che risultano così più difficilmente passivabile, v. Fig. 10.8. Si ricorda che il tenore di Cr libero nell'inox non deve scendere al di sotto del 12 %, se si vuole garantire la formazione di un film compatto e protettivo. La sensibilizzazione degli acciai inox è un fenomeno frequentemente collegato con i processi di saldatura e può essere in genere evitato con l'uso di acciai a basso contenuto di carbonio (ad es. AISI 304L, 316L, L=low carbon) oppure di acciai stabilizzati contenenti in lega piccole quantità di titanio o niobio (AISI 321 al Ti, AISI 347 al Nb), in cui durante un trattamento termico di stabilizzazione si induce la precipitazione omogenea nella struttura di TiC o NbC, sottraendo carbonio dalla matrice ed impedendo la precipitazione di carburi di cromo durante saldatura.
La c. sotto sforzo è un tipo di corrosione localizzata che si realizza con la formazione di cricche assai sottili in seguito all'azione combinata di un mezzo corrosivo di blanda aggressività, ma specifico per un dato materiale metallico, e di uno stato di sollecitazione di trazione di entità anche non rilevante. Il fenomeno è particolarmente pericoloso in quanto può dar luogo ad improvvisi cedimenti del materiale, senza che questo sia visibilmente deteriorato. La SCC colpisce una vasta gamma di materiali metallici come gli acciai al carbonio, gli acciai inossidabili, le leghe leggere, quelle di rame e titanio, ecc., sotto l'azione di ambienti di natura alquanto differenziata (alogenuri, nitrati, solfuri, soluzioni caustiche ecc.). Data la varietà delle combinazioni possibili, non è possibile parlare di materiali immuni o suscettibili di SCC, o di ambienti promotori oppure no. Per l'innesco della SCC devono comunque verificarsi le seguenti condizioni:
- è necessaria la presenza di tensioni di trazione. Tali tensioni possono derivare oltre che da carichi esterni
applicati, anche da stati autotensionali causati da montaggi forzati, dilatazioni termiche o cedimento di
vincoli;
- l'attacco si produce solo su leghe metalliche e, generalmente, non su metalli puri.
- l'attacco si produce solo per specifiche combinazioni ambiente - M.M.
- la presenza di alcuni elementi di lega può aumentare la suscettibilità di un metallo o determinarne l'immunità. Ad es;, la presenza di azoto negli acciai inox aumenta la suscettibilità alla SCC in MgCl2 bollente. La presenza di Si (1,5 %) diminuisce la sensibilità di certi ottoni in ambienti ammoniacali.
La SCC si realizza con formazione di cricche orientate perpendicolarmente alla direzione della tensione meccanica applicata. Queste possono essere inter/trans-granulari, seguendo comunque una o più direzioni principali, con piccole ramificazioni assai caratteristiche, vedi Fig. 10.9.
Sotto l'aspetto fenomenologico l'attacco si sviluppa sempre attraverso un innesco di cricca per pitting, per poi propagarsi all’interno del materiale sotto l'azione combinata delle tensioni di trazione e dell’ambiente corrosivo. Una volta iniziato, il danneggiamento procede all'interno del materiale con un meccanismo complesso (non del tutto chiarito) che comporta stadi susseguenti di dissoluzione elettrochimica all'apice della cricca e di frattura meccanica. L'instaurarsi di coppie galvanicche tra apice della cricca (area anodica) e pareti della stessa (area catodica) e il possibile intervento di fenomeni infragilenti (ad es. in materiali suscettbili all’infragilimento da idrogeno) sul fronte di avanzamento della cricca, possono dar luogo a velocità di avanzamento anche estremamente alte (mm/ora).
I casi più comuni di SCC si riferiscono agli acciai al C in presenza di nitrati (industria dei fertilizzanti), cianuri, solfuri, soluzioni caustiche concentrate (infragilimento caustico). Gli acciai inox sono suscettibili in ambienti contenenti cloruri o solfuri. Il rame e sue leghe possono criccarsi velocemente in presenza di sali ammoniacali o di mercurio. In Tab. 10.6 è riportato il comportamento a S.C.C. di alcuni materiali metallici.
I metodi di prevenzione della SCC consistono nella scelta di materiali non suscettibili di cracking negli ambienti in cui si impiegano, nell'evitare inquinamenti pericolosi, nell'eliminare la presenza di sollecitazioni meccaniche in fase di montaggio o servizio e nell'evitare tensioni di saldatura. Possono rivelarsi utili anche trattamenti di pallinatura e di protezione catodica.
Uno dei metodi più diffusi ed efficaci per la protezione dei materiali metallici dal degrado ambientale è quello di applicare sulla loro superficie film o strati resistenti alla corrosione, in modo da isolare fisicamente (protezione passiva) il materiale metallico dall’ambiente aggressivo circostante (placcature metalliche, smalti, rivestimenti organici a spessore, pitture e vernici). L’efficacia della protezione passiva è però funzione dell’integrità del ricoprimento. Spesso, pertanto, si impiegano strati capaci di proteggere anche per inibizione o per protezione catodica il materiale (protezione mista). Possono esserci anche altre finalità del ricoprimento, come ad esempio un fattore estetico o una azione biocida.
Possiamo distinguere in: placcatura, incamiciatura e rivestimenti sottili (da deposito) formati in loco. La placcatura di materiali resistenti a corrosione su materiali resistenti meccanicamente viene realizzata per colaminazione o per riporto di saldatura, e comunque realizzando in tal modo una continuità cristallografica e un legame metallurgico tra i diversi materiali. Vi sono problemi per la definizione dei trattamenti termici da condurre sull’intero manufatto, così come accorgimenti da adottare per le saldature di ripristino. Quando non vi sono particolari requisiti di servizio (problemi di scambio termico o alte pressioni) si può ricorrere ad una semplice incamiciatura, saldata per punti sull’apparecchio già costruito. Questi metodi richiedono una quantità importante di materiale pregiato, sono costosi e possono impiegarsi per apparecchiature di forma geometrica semplice.
Più economici e di semplice applicazione, da impiegarsi quindi a pezzi di geometria più complessa, sono i rivestimenti da deposito. Bisogna siano spessi, compatti ed aderenti al substrato per essere protettivi, ed inoltre è bene che siano anodici rispetto al materiale da proteggere, in modo da offrire al substrato anche protezione catodica.
I metodi più usati sono:
Altre tecniche sono la metallizzazione sottovuoto (vapor deposition), cromizzazione (cromo) e colorizzazione (alluminio) e la deposizione a spruzzo (flame/plasma spraying).
La deposizione a caldo si applica soprattutto sugli acciai, applicando rivestimenti di metalli a basso punto di fusione (Zn, Pb, Sn, Al). I problemi che si incontrano sono, a parte una preparazione adeguata della superficie, nella bagnabilità del substrato ad opera del bagno fuso (hot dipping, procedimento per immersione). Nel caso dello zinco su acciaio vi è una certa solubilità reciproca, e pertanto si verifica la formazione di leghe ferro-zinco. Gli strati normalmente depositati sono dello spessore di qualche decina di micrometri.
La deposizione galvanica, o elettrodeposizione, consiste nell’immergere il pezzo da rivestire in una soluzione contenente il metallo da depositare, e facendo passare corrente tra il pezzo e un elettrodo di lavoro. In funzione del metallo da depositare e delle condizioni operative adottate (temperatura, composizione del bagno, tempo e additivi specifici) si possono ottenere film sottili di diversi micrometri o anche spessi fino a qualche mm. Lo strato superficiale, può essere composto da un singolo ricoprimento o da più strati. Per esempio, i paraurti cromati (di qualità) che si usavano per le automobili avevano un primo strato di rame (per l’adesione) uno secondo di nichel (protezione dalla corrosione) e uno finale di cromo (estetico).
La scelta tra l’elettrodeposizione e il metallo di immersione dipende molto dall’impiego del manufatto. In generale, se sono richiesti depositi coprenti di piccolo spessore, si sceglie l’elettrodeposizione, in quanto è più facile dosare il materiale di placcatura (e dunque se ne consuma meno) e la finitura superficiale è in genere buona. Per esempio, nelle confezioni alimentari, per sostanze poco aggressive (caffè e tabacco), vanno bene film sottili (elettrodeposizione), per elementi più aggressivi (frutta e pesce) o per scatole da inviare in zone tropicali (corrosione filiforme) occorre aumentare lo spessore e si sceglie un deposito più spesso ottenuto per immersione a caldo.
La deposizione chimica (electroless)sfrutta una reazione chimica superficiale per produrre il rivestimento desiderato. L’esempio classico dello spostamento del rame da una soluzione contenente suoi ioni con deposizione sulla superficie di compomente in ferro è istruttivo. A livello industriale, comunque, si impiegano riducenti come l’idrazina, ipofosfiti per accelerare la riduzione dello ione metallico desiderato. In tal modo non è necessario che abbia luogo contemporaneamente un processo di dissoluzione anodica del metallo da rivestire e la reazione procede finchè nel bagno c’è traccia del riducente additivato. In tal modo si possono depositare una grande varietà di metalli, anche su substrati non metallici (argentatura degli specchi). I bagni electroless di questo tipo sono in genere complesssi (brillantanti, emulsionanti, stabilizzanti ecc..).
SMALTI: Gli smalti impiegati nell’industria chimica hanno composizioni particolari in funzione della specifica applicazione (acido-resistenti con composti del boro e con ossidi di titanio e zirconio). Per le applicazioni industriali devono avere coefficienti di dilatazione termica simili al substrato metallico, buona resistenza agli shock termici e buona adesione al metallo (attenzione allo sviluppo di H2; operare con inibitori che limitino la corrosione del metallo e a caldo, per non disciogliere grandi quantità di idrogeno). Problema della difettosità e delle scagliature (difficile riparazione).
STRATI A SPESSORE: I rivestimenti protettivi a spessore, con materiali quali ebanite, gomme, materiali polimerici e bituminosi, hanno un largo impiego nell’industria, e sono utilizzati anche per la protezione di strutture sotterranee e sottomarine. I rivestimenti si effettuano di norma in fabbrica mediante coestrusione, sinterizzazione di polveri ecc.., ma è possibile, per alcuni casi, realizzarli anche in loco mediante applicazione sotto forma di nastri adesivi).
I problemi sono: l’adesione con il substrato metallico, l’invecchiamento e l’infragilimento del rivestimento nel tempo. Nelle applicazioni industriali, bisogna inoltre tenere conto del limitato scambio termico e della relativamente bassa temperatura massima di impiego (max 70 ¸ 80°C).
La distinzione tra vernici e pitture, è nella presenza, in queste ultime, dei pigmenti. La vernice, ha in genere solo una funzione ricoprente, fornendo una protezione di tipo passivo; l’impiego di pigmenti, oltre alla colorazione, può essere specifica di protezione (catodica, anodica o passivante) per cui vanno scelti in modo appropriato. Un normale rivestimento con pittura viene condotto con una o più mani di fondo, con strati che aderiscono bene al substrato (importante è dunque la preparazione della superficie da proteggere: sgrassaggio, decapaggio e sabbiatura) e che svolgano una funzione attiva di protezione, ed uno o più strati di finitura, con l’intenzione di isolare lo strato di fondo (spesso sensibile all’umidità) dall’ambiente esterno.
La natura del veicolo, può essere molto diversa e classificabile, ad esempio, in base al meccanismo di essiccamento, che può avvenire per ossidazione-polimerizzazione (vernici a base di olio di lino, resine alchidiche epossidiche e poliesteri) e per evaporazione (nitrato di cellulosa) oppure per semplice raffreddamento da alta temperatura (vernici con legante catramoso o bituminoso). Vernici a base di acqua sono impiegate nell’industria automobilistica nei bagni elettroforetici.
Molto importante è l’azione dei pigmenti che, in buona parte, svolgono un’azione passivante, in grado cioè di promuovere e mantenere lo strato di passività sul metallo (ad esempio il minio 2PbO-PbO2) o i cromati di Zn e Pb. Come protezione catodica può essere utilizzato lo Zn (percentuali 50 %) (pigmenti catodici).
Sono film superficiali protettivi formati in situ per via chimica o elettrochimica. Possono servire tal quali o come base per vernici. Sono molto impiegati per gli acciai, per l’alluminio e per lo zinco. I più usati sono l’ossidazione anodica, la fosfatizzazione e la cromatazione.
L’OSSIDAZIONE ANODICA è il metodo più diffuso per la protezione dell’alluminio. Consiste per formare per via elettrochimica un film superficiale di ossido di spessore ragguardevole (3 ¸ 30 mm) di buona compattezza e stabilità. Si esegue in due tempi: in una prima fase si induce la formazione e l’accrescimento del film per via elettrochimica impiegando bagni opportuni (ossalico, borico, bicromato e più comunemente solforico) ed eventualmente aggiungendo coloranti, mentre per immersione in acqua bollente, o esposizione al vapore a T = 100 ¸ 150°C, si idrata, si sigilla e si stabilizza il film conferendogli le specifiche caratteristiche protettive ed estetiche.
LA CROMATAZIONE è un processo per la protezione di materiali non ferrosi quali Zn e Cd. Questi metalli, quando usati nella protezione come anodi sacrificali, possono, a seconda dell’ambiente corrosivo, corrodersi velocemente (ambiente marino), e quindi richiedere a loro volta una protezione. Il trattamento consiste nell’immergere il componente in una soluzione acida di cromato, con formazione di prodotti superficiali con caratteristiche protettive. Nel caso dello Zn, il film è spesso pochi micron, trasparente e di colore dal giallo a verde scuro.
LA FOSFATAZIONE consiste nel formare uno strato superficiale sul metallo (in genere ferro) di fosfati misti di Zn, Mn e Fe, fortemente aderente al substrato e ottimo come base per pitture e vernici. L’azione protettiva si esplica nella protezione del materiali nei punti dove la vernice o lo stato organico si sono lesionati a causa della penetrazione dell’umidità, garantendo la presenza del fosfato (inibitore) e inibendo la formazione di focolai corrosivi. Il processo consiste nel trattamento in bagno di acido fosforico e fosfati acidi di Zn e Mn. La reazione con il ferro, con formazione di fosfato primario FeH2PO4 e sviluppo di idrogeno, con consumo di H3PO4, sposta gli equilibri di idrolisi degli altri sali e causa precipitazione sulla superficie. Spesso si impiegano temperature di 50 70°C e acceleranti (ossidanti NO3- ClO3- e H2O2) per sostituire la riduzione di H3O+ e realizzare il processo. Altri impieghi sono la protezione temporanea di componenti metallici da immagazzinare (spessore di fosfato minori di 100mm impiegati come base per successiva pitturazione) oppure il pretrattamento superficiale alle operazioni di formatura di pezzi metallici (fisso il lubrificante, ad esempio stearati ® estrusione, trafilatura).
Sono criteri basati essenzialmente sulla conoscenza di base dei fenomeni corrosivi. Per quanto riguarda l'aspetto termodinamico si ricorda l'impiego dei diagrammi di Pourbaix, che permettono di prevedere sia lo stato di attività del materiale, che la precipitazione di composti capaci di passivare la superficie del metallo. Parallelamente la conoscenza degli aspetti CINETICI (curve tensione-corrente) dà utili informazioni sulla localizzazione dei fenomeni dissipativi e permette quindi di indirizzarsi al controllo dei processi più importanti.
Infine la conoscenza di base permette di non commettere errori banali quali potrebbero essere accoppiare materiali diversi o impiegare acciai inox in ambienti riducenti o contenenti cloruri ed infine, permette di selezionare i mezzi di prevenzione e controllo più efficaci per quelle specifiche problematiche.
Questi criteri di selezione si basano sulla disponibilità di dati e informazioni di carattere sperimentale sull'impiego dei materiali nei vari ambienti aggressivi. Un primo orientamento pratico è fornito dal panorama di combinazioni naturali metallo-mezzo corrosivo già collaudate dall'esperienza, le più importanti delle quali sono:
- piombo ---- soluzioni di acido solforico;
- acciaio --- acido solforico concentrato;
- acciai inox --- soluzioni acido nitrico;
- Monel (66Ni-31Cu) --- acido fluoridrico;
- Hastelloy (Ni-Cr-Mo) --- acido cloridrico;
- alluminio --- atmosfere varie;
- stagno --- acqua distillata e mezzi alimentari
- titanio --- mezzi fortemente ossidanti.
Esistono tuttavia banche dati su cui cercare dati più specifici; le più utilizzate sono:
1-CORROSION DATA SURVEY.
Riporta il comportamento corrosionistico dei più comuni materiali metallici e non, al variare della temperatura e concentrazione delle soluzioni aggressive; per ambienti acidi (solforico, nitrico, cloridrico, fosforico) fornisce diagrammi come da Figg. .
2-CORROSION GUIDE di E.RABALD.
Riporta i dati di corrosione di moltissimi materiali esposti a 450 ambienti corrosivi diversi in svariate condizioni di temperatura e concentrazione. Il tasso di corrosione massimo (accettabile) viene espresso in relazione alla classe di costo dei materiali. Come esempio, nella Fig. viene mostrato quanto reperibile per materiali esposti in ambienti contenenti acido ossalico.
3-DECHEMA WERSCKOFFE TABELLEN.
Simile al precedente, ora in lingua inglese nella denominazione "Corrosion Data Sheets".
Fonte: http://www1.diccism.unipi.it/De_Sanctis_Massimo/Slide%20lezioni%20-%20Materiali%20%202013/elementi%20di%20corrosione.doc
Sito web da visitare: http://www1.diccism.unipi.it
Autore del testo: non indicato nel documento di origine
Il testo è di proprietà dei rispettivi autori che ringraziamo per l'opportunità che ci danno di far conoscere gratuitamente i loro testi per finalità illustrative e didattiche. Se siete gli autori del testo e siete interessati a richiedere la rimozione del testo o l'inserimento di altre informazioni inviateci un e-mail dopo le opportune verifiche soddisferemo la vostra richiesta nel più breve tempo possibile.
I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
"Ciò che sappiamo è una goccia, ciò che ignoriamo un oceano!" Isaac Newton. Essendo impossibile tenere a mente l'enorme quantità di informazioni, l'importante è sapere dove ritrovare l'informazione quando questa serve. U. Eco
www.riassuntini.com dove ritrovare l'informazione quando questa serve